Clear Sky Science · fr
Découverte de l’hydroxytriazole comme inhibiteur potentiel de la glyoxalase‑I grâce à des techniques de conception assistée par ordinateur
Pourquoi bloquer un petit agent nettoyeur cellulaire pourrait combattre le cancer
Les cellules cancéreuses se développent souvent si rapidement qu’elles s’ensevelissent dans leurs propres déchets. L’un de leurs stratagèmes de survie est une équipe de nettoyage intégrée qui neutralise les sous‑produits nocifs de la combustion du sucre. Cette étude explore comment désactiver un membre clé de cette équipe, une enzyme appelée glyoxalase‑I, en utilisant l’informatique pour criblere des dizaines de milliers de molécules et des expériences pour tester les meilleures candidates. L’objectif est de découvrir de nouveaux points de départ pharmacologiques qui pourraient un jour aider les médecins à empoisonner sélectivement les cellules cancéreuses de l’intérieur.
Un système caché d’élimination des déchets à l’intérieur de nos cellules
Chaque cellule décompose constamment le sucre pour produire de l’énergie, et ce processus génère un déchet réactif appelé méthylglyoxal. À des niveaux normaux, notre organisme convertit le méthylglyoxal en acide lactique inoffensif via le système glyoxalase, une voie en deux étapes qui dépend de la molécule aide glutathion. La glyoxalase‑I est la première et la plus cruciale étape de cette chaîne. Les cellules cancéreuses, qui brûlent le sucre à un rythme effréné, dépendent fortement de la glyoxalase‑I pour empêcher le méthylglyoxal d’atteindre des niveaux toxiques. Si cette enzyme est bloquée, le méthylglyoxal s’accumule et peut pousser les cellules endommagées vers la mort programmée. Cela fait de la glyoxalase‑I une cible attirante pour des médicaments anticancéreux qui s’attaquent à une faiblesse fondamentale du métabolisme tumoral.
Explorer l’espace chimique avec le silicium et les statistiques
Plutôt que de tester des substances au hasard en laboratoire, les chercheurs ont utilisé la conception de médicaments assistée par ordinateur pour parcourir une grande collection commerciale de plus de 50 000 petites molécules. Un logiciel spécialisé a d’abord nettoyé et standardisé chaque molécule, puis prédit sa forme 3D et son comportement à un pH proche de celui du corps. Une étape de criblage virtuel rapide a évalué l’aptitude de chaque candidat à s’insérer dans le site actif de la glyoxalase‑I. L’équipe a ensuite appliqué des règles simples concernant la taille, la solubilité et d’autres propriétés de « drug‑likeness » pour écarter les molécules peu susceptibles de fonctionner dans l’organisme. Un programme de docking plus détaillé a examiné comment les molécules les plus prometteuses pourraient s’orienter à l’intérieur de l’enzyme, en particulier comment elles pourraient atteindre et saisir l’atome de zinc qui se trouve au cœur de la chimie de la glyoxalase‑I.
Une nouvelle façon d’attraper le noyau métallique de l’enzyme
Les efforts antérieurs pour bloquer la glyoxalase‑I se concentraient sur des groupements chimiques bien connus, tels que les acides carboxyliques et les acides hydroxamiques, qui lient bien les métaux mais souffrent souvent d’une faible stabilité ou d’effets indésirables. La présente étude a révélé à la place un type différent d’unité « accroche‑métal » : un noyau hydroxytriazole. Parmi seize molécules mieux classées choisies pour l’achat et les tests en laboratoire, l’une portant ce noyau — codée SPB07393SC — s’est détachée. Dans le docking virtuel, son groupe hydroxytriazole atteignait l’atome de zinc, tandis que ses deux cycles aromatiques se logeaient dans des poches lipophiles voisines de l’enzyme. Des simulations informatiques du complexe sur des dizaines de nanosecondes ont suggéré que la molécule restait fermement liée, avec des distances stables, une conformation protéique compacte et un réseau persistant de liaisons hydrogène.
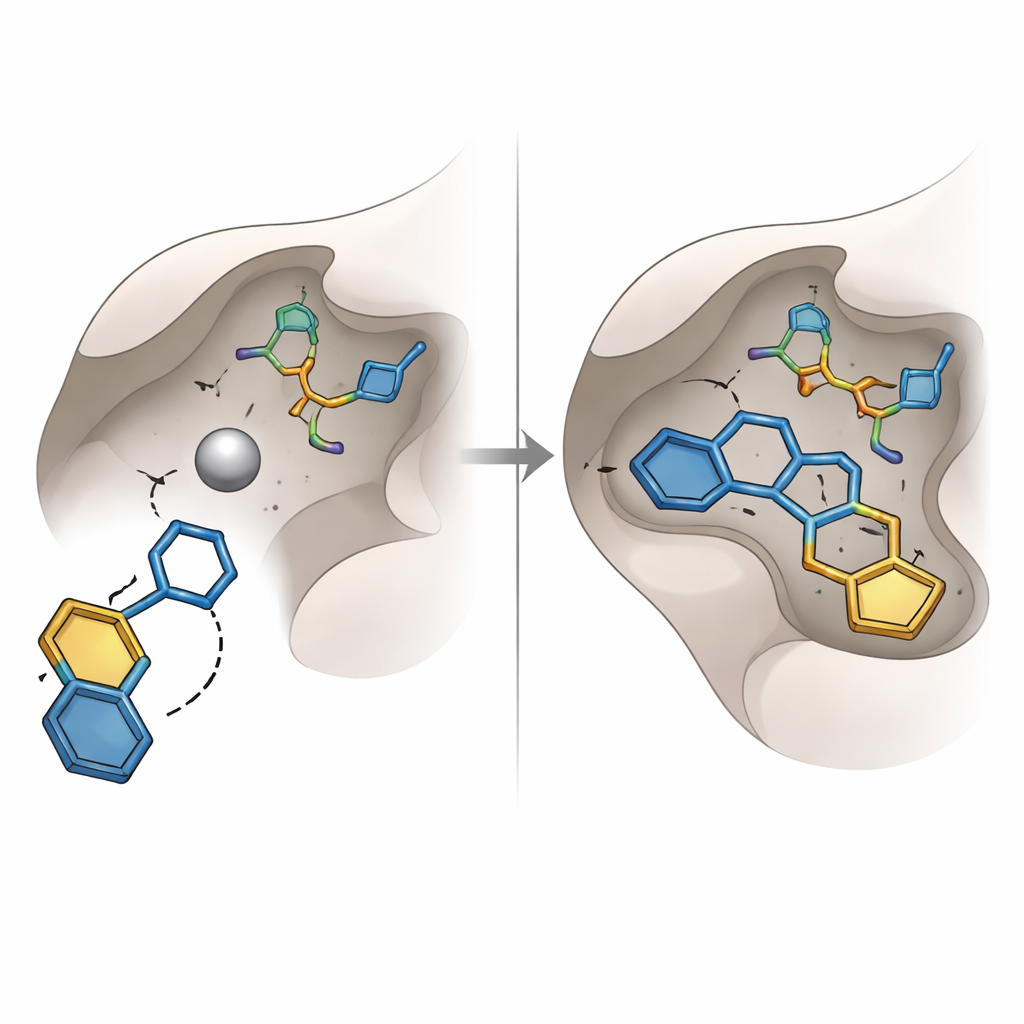
Mettre les prédictions à l’épreuve
Pour savoir si les modèles informatiques se traduisaient par des effets réels, l’équipe a mesuré dans quelle mesure les molécules sélectionnées ralentissaient l’activité de la glyoxalase‑I humaine purifiée dans un test en microplaques. Quinze des seize candidates n’ont montré qu’une inhibition faible ou négligeable dans les conditions testées, soulignant les pièges de la dépendance exclusive aux scores de docking statiques. En revanche, SPB07393SC a fortement inhibé l’enzyme, avec une puissance en micromolaire moyen qui en fait un « hit » précoce solide plutôt qu’un médicament fini. Des outils logiciels supplémentaires ont prédit que cette molécule devrait présenter une solubilité acceptable, une bonne absorption, la capacité d’atteindre le cerveau si nécessaire, et une faible probabilité d’induction de certaines toxicités génétiques ou hépatiques, bien que ces prédictions de sécurité nécessitent encore une confirmation expérimentale.
Ce que cela signifie pour les futurs médicaments anticancéreux
Ce travail introduit l’hydroxytriazole comme une nouvelle façon d’ancrer des candidats‑médicaments sur l’atome de zinc au cœur de la glyoxalase‑I, élargissant le répertoire de tours chimiques à la disposition des concepteurs de médicaments. Si SPB07393SC n’est lui‑même qu’un point de départ, sa combinaison de capacité à bloquer l’enzyme, de propriétés physico‑chimiques prédites favorables et d’un ancrage stable dans des simulations dynamiques en fait une ossature prometteuse pour un affinage ultérieur. Plus largement, l’étude illustre à la fois les forces et les limites du criblage guidé par ordinateur : il peut rapidement réduire d’immenses bibliothèques chimiques à quelques candidats réalistes, mais des expérimentations rigoureuses en laboratoire restent essentielles pour révéler quelles molécules désactivent réellement l’enzyme dont dépendent les cellules cancéreuses pour gérer leurs déchets toxiques.
Citation: Al-Qazzan, M., Al-Balas, Q., Alnajjar, B. et al. Discovery of hydroxytriazole as a potential glyoxalase-I inhibitor utilizing computer-aided drug design techniques. Sci Rep 16, 9945 (2026). https://doi.org/10.1038/s41598-026-40497-4
Mots-clés: glyoxalase I, métabolisme du cancer, conception de médicaments assistée par ordinateur, inhibiteurs liant le zinc, dockage moléculaire