Clear Sky Science · fr
DPAS : score d’anomalie des peptides associés aux maladies pour identifier des peptides pathogènes via l’apprentissage à classe unique
Pourquoi de petits fragments de protéines comptent pour notre santé
Les peptides — courtes séquences de protéines — sont devenus des acteurs majeurs de la médecine moderne. Ils peuvent servir de messagers précis dans l’organisme et sont de plus en plus utilisés comme médicaments et marqueurs de maladie. Pourtant, déterminer quels peptides sont réellement liés à la maladie repose souvent sur des exemples clairs de peptides « malades » et « non‑malades », ce que la biologie fournit rarement. Cette étude présente une nouvelle façon de repérer des peptides potentiellement nocifs en n’utilisant que ceux que l’on sait déjà impliqués dans la maladie, offrant une voie plus rapide et moins biaisée pour découvrir de futurs diagnostics et traitements.
Le défi de trouver le groupe « non‑malade »
Les modèles informatiques traditionnels apprennent en comparant deux camps : des exemples positifs connus pour être liés à la maladie et des exemples négatifs supposés inoffensifs. En recherche sur les peptides, ce deuxième groupe pose problème. Beaucoup de peptides n’ont simplement pas été testés, donc les étiqueter comme « non‑malades » peut être trompeur et introduire des biais. Des études antérieures sur des peptides anti‑cancer ou anti‑inflammatoires ont atteint des précisions impressionnantes, mais ont souvent reposé sur des jeux de données négatifs construits manuellement ou supposés. De ce fait, leurs modèles peuvent échouer à détecter des signaux rares ou de nouveaux types de peptides pathogènes qui ne ressemblent pas aux données d’entraînement.
Apprendre de ce que l’on sait, plutôt que de ce que l’on suppose
Les auteurs empruntent une voie différente : au lieu de forcer un problème bipolaire, ils considèrent les peptides associés à la maladie comme un groupe cohérent et se demandent « À quoi ressemble ce groupe en détail ? ». Ils rassemblent plus de 760 000 peptides humains mutés issus d’une base de données spécialisée en cancer et décrivent chaque peptide par un ensemble riche de caractéristiques. Celles‑ci incluent la fréquence d’apparition de chaque acide aminé, l’arrangement des paires d’acides aminés, des traits physico‑chimiques de base comme le volume et l’affinité pour l’eau, et de courtes séquences récurrentes appelées motifs. Une technique appelée analyse en composantes principales compresse ensuite cette description haute dimension en une forme plus maniable tout en préservant les principales sources de variation.
Repérer les peptides inhabituels avec des modèles à classe unique
Avec cet espace de caractéristiques compressé, l’équipe entraîne trois modèles « à classe unique » — des algorithmes conçus pour apprendre la forme d’un seul groupe et signaler tout ce qui n’y correspond pas. Ils testent des machines à vecteurs de support à classe unique (One‑Class SVM), des Isolation Forests et un type de réseau de neurones appelé autoencodeur. L’autoencodeur apprend à comprimer les caractéristiques de chaque peptide dans une représentation interne étroite puis à les reconstruire ; les peptides appartenant au motif de la maladie appris sont reconstruits avec précision, tandis que les peptides inhabituels entraînent une plus grande erreur de reconstruction. La comparaison des scores d’anomalie normalisés pour toutes les méthodes montre que l’autoencodeur produit le regroupement le plus serré des peptides typiques et la séparation la plus nette entre éléments internes et externes. En fixant un seuil sur l’erreur de reconstruction autour du 95e percentile, le modèle classe la majorité des peptides comme probablement associés à la maladie tout en signalant de manière cohérente une petite fraction comme atypique.
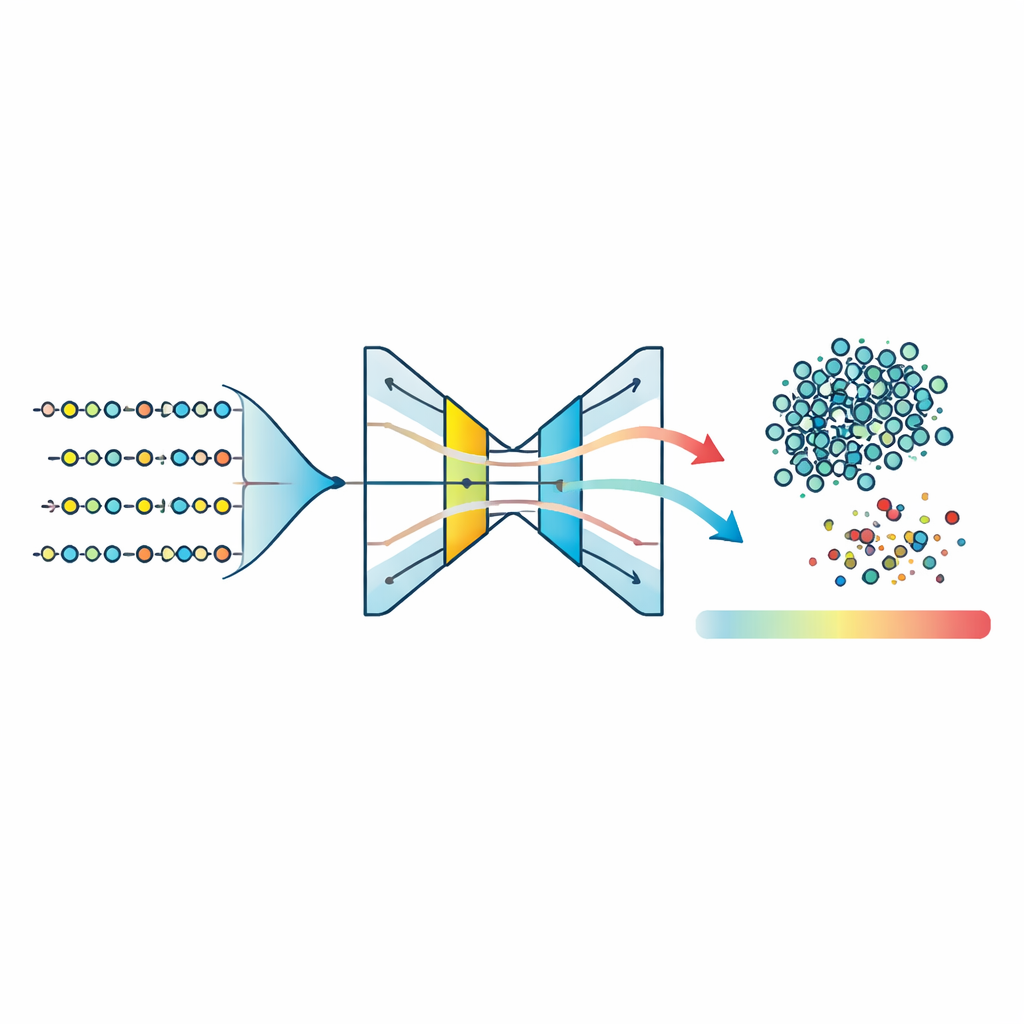
Transformer des scores complexes en un seul nombre significatif
Pour rendre les résultats plus faciles à interpréter biologiquement, les auteurs introduisent le score d’anomalie des peptides liés à la maladie (DPAS). Ce score combine deux ingrédients : à quel point un peptide apparaît inhabituel pour l’autoencodeur (son erreur de reconstruction normalisée) et dans quelle mesure ses caractéristiques contribuent aux prédictions, mesurées par une méthode d’explicabilité populaire appelée SHAP. En pratique, les motifs et certains traits physico‑chimiques se révèlent particulièrement informatifs. DPAS combine ces signaux de sorte que les peptides à la fois structurellement étranges et soutenus par des caractéristiques biologiquement pertinentes reçoivent des rangs plus élevés. Les peptides arrivant en tête sont ensuite examinés avec un outil de recherche de motifs qui les relie à des signatures fonctionnelles connues telles que des sites de phosphorylation, des régions de liaison aux métaux et d’autres motifs de régulation fréquemment impliqués dans la signalisation et le contrôle enzymatique.
Ce que cela signifie pour les diagnostics et médicaments futurs
Concrètement, ce travail propose un filtre plus intelligent pour repérer des peptides suspects sans prétendre connaître ceux qui sont définitivement inoffensifs. En apprenant uniquement à partir d’exemples confirmés liés à la maladie puis en classant de nouveaux candidats avec DPAS, les chercheurs peuvent prioriser une courte liste biologiquement plausible de peptides pour des tests en laboratoire. Beaucoup des candidats les mieux classés contiennent des motifs fonctionnels bien connus, renforçant l’idée qu’ils peuvent jouer des rôles dans des processus pathologiques. Bien que la méthode repose encore sur des hypothèses et manque de peptides « sûrs » prouvés expérimentalement pour une validation complète, elle fournit une base plus réaliste et transparente pour la découverte de biomarqueurs peptidiques et pourrait être adaptée à d’autres types de données biologiques où les exemples négatifs fiables sont rares.
Citation: Khalid, Z., Khalid, R. & Sezerman, O.U. DPAS: disease-associated peptide anomaly score for identifying pathogenic peptides via one-class learning. Sci Rep 16, 9170 (2026). https://doi.org/10.1038/s41598-026-40099-0
Mots-clés: peptides associés aux maladies, détection d’anomalies, autoencodeur, découverte de biomarqueurs, apprentissage à classe unique