Clear Sky Science · fr
Génération de novo et criblage in silico de candidats peptides antidiabétiques via un cadre d’apprentissage profond–attention avec fusion de caractéristiques physico-chimiques
Pourquoi une conception de peptides plus intelligente compte pour le diabète
Le diabète touche des centaines de millions de personnes dans le monde et les médicaments actuels ne fonctionnent pas parfaitement pour tous. Beaucoup de traitements perdent en efficacité au fil du temps ou provoquent des effets secondaires. Une option prometteuse est une classe de petites protéines appelées peptides antidiabétiques, qui peuvent ajuster la glycémie avec une grande précision. Le problème est que découvrir de nouveaux médicaments peptidiques en laboratoire est lent et coûteux. Cette étude présente une chaîne d’outils pilotée par ordinateur qui peut inventer et trier un grand nombre de peptides candidats antidiabétiques, guidant les chercheurs vers les candidats les plus prometteurs à tester en situation réelle.
Des peptides connus pour le diabète à des données de départ propres
Les chercheurs ont commencé par assembler une collection de haute qualité de peptides montrés expérimentalement comme influant sur la glycémie, principalement en modulant des hormones telles que le GLP-1 ou des enzymes comme la DPP‑IV. Ceux-ci ont constitué les exemples « positifs ». Ils ont ensuite construit un ensemble « négatif » correspondant, composé de peptides sans activité antidiabétique rapportée, choisis avec soin pour que la longueur, la composition et la chimie de base ressemblent à celles des positifs. Pour éviter de tromper le modèle avec des quasi‑doublons, ils ont utilisé des outils de similarité de séquence afin de s’assurer que des peptides étroitement apparentés n’apparaissent jamais à la fois dans les groupes d’entraînement et de test. Cette séparation consciente de l’homologie garantissait que le système soit évalué sur sa capacité à reconnaître de vrais nouveaux motifs plutôt qu’à mémoriser d’anciens.
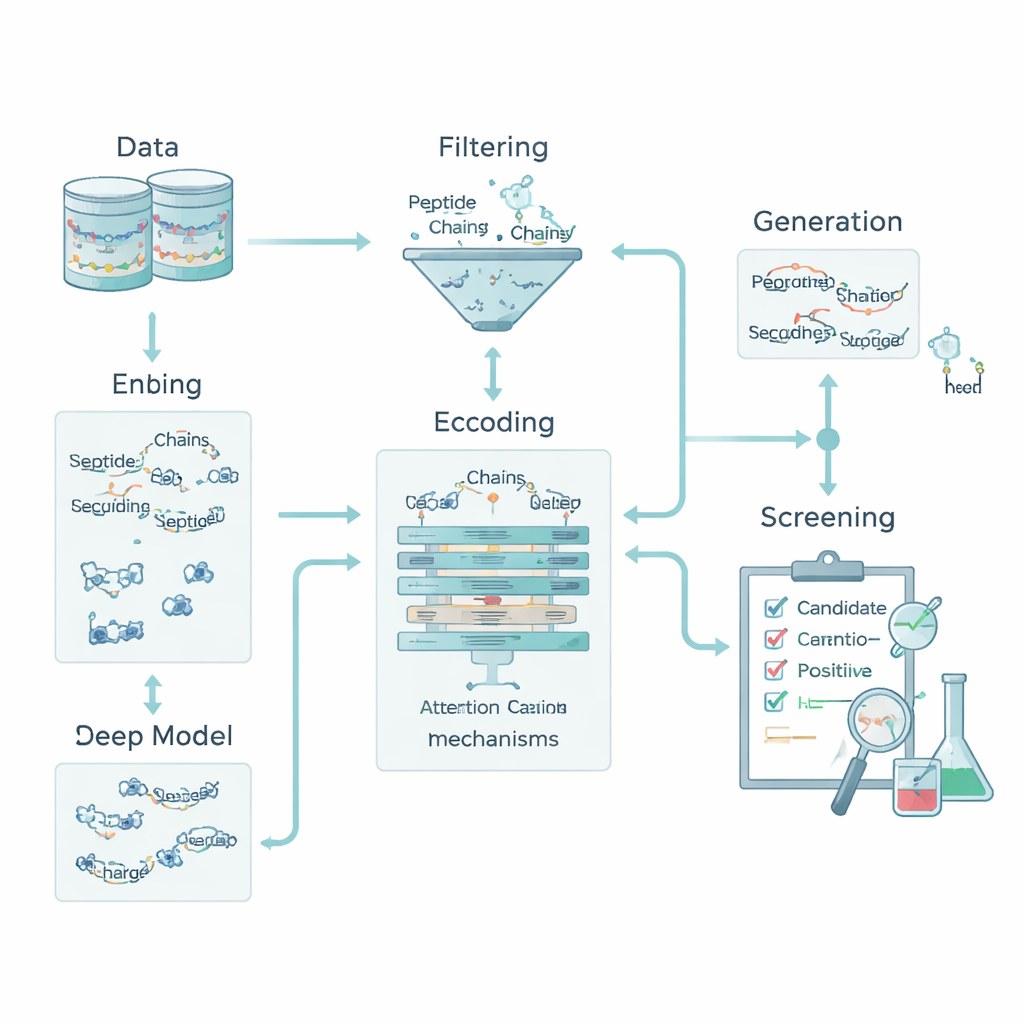
Encoder la chimie pour que les machines lisent les peptides
Pour un ordinateur, un peptide n’est qu’une suite de lettres représentant des acides aminés. Pour relier ces lettres à la biologie, l’équipe a transformé chaque acide aminé en cinq traits chimiques de base : son caractère hydrophobe, sa charge électrique, sa tendance à former des liaisons hydrogène, sa masse et la présence éventuelle d’un anneau aromatique. Cela a converti chaque peptide en une petite « image » capturant à la fois l’ordre et la chimie. Par-dessus cela, ils ont ajouté des descripteurs à l’échelle du peptide, tels que la charge globale, l’hydrophobicité moyenne et l’indice de Boman, qui renseigne sur la propension d’un peptide à se lier à d’autres protéines. Ensemble, ces caractéristiques permettent au modèle d’examiner à la fois des motifs locaux — de courts motifs d’acides aminés — et des propriétés globales qui influencent le comportement d’un peptide dans l’organisme.
Un moteur d’apprentissage profond qui explique ses choix
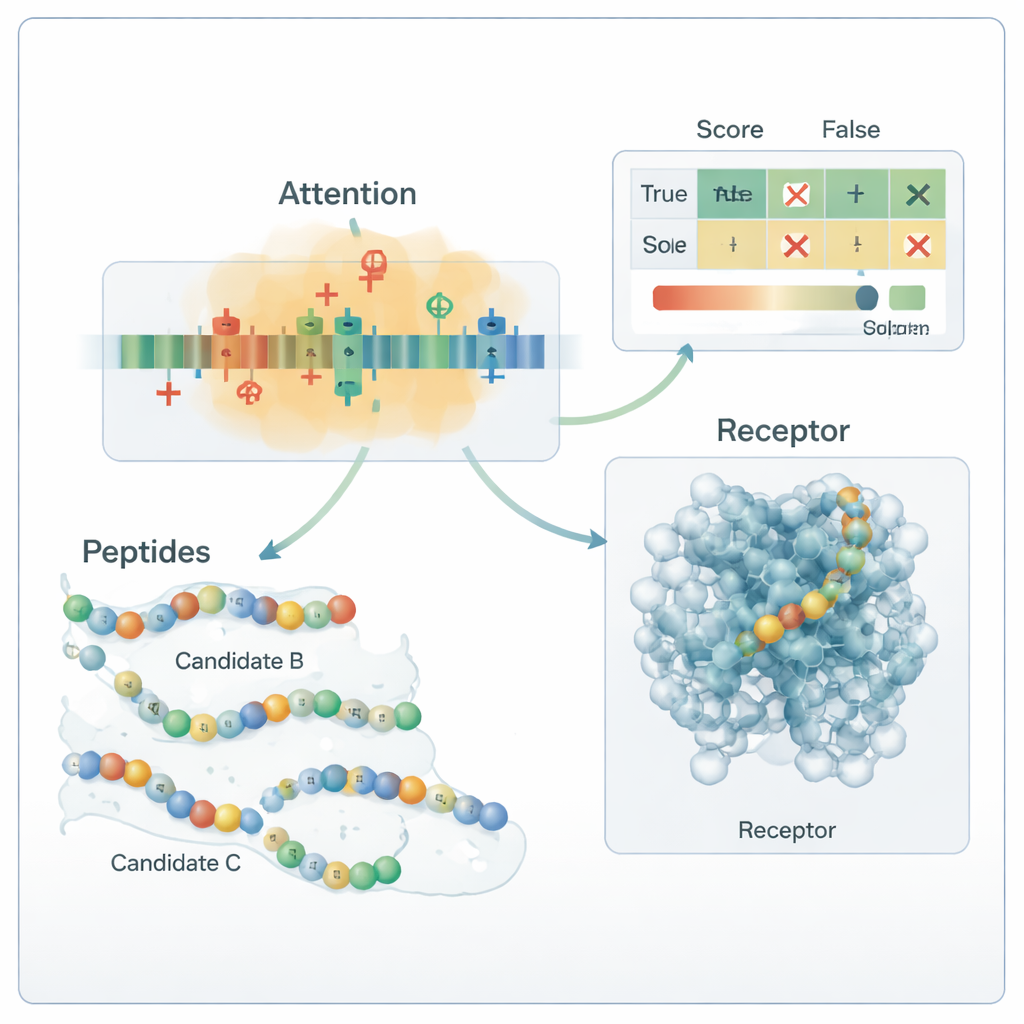
Le cœur de la chaîne est un modèle hybride d’apprentissage profond. Un réseau de neurones convolutionnel (CNN) parcourt le peptide à la recherche de courts motifs qui tendent à apparaître dans les peptides actifs, un peu comme des filtres dans un système de reconnaissance d’images. Au‑dessus, une couche d’attention apprend quelles positions dans la séquence sont les plus importantes, capturant des relations à longue portée entre des résidus distants. La sortie de ce moteur de séquence est fusionnée avec les descripteurs chimiques globaux et transmise à plusieurs classifieurs d’apprentissage automatique classiques — machines à vecteurs de support, arbres de décision, k plus proches voisins et arbres à gradient boosté. Une méthode d’optimisation spécialisée, appelée OptimizedTPE, ajuste automatiquement leurs paramètres, trouvant un équilibre entre précision et risque de surapprentissage. Le mécanisme d’attention fournit aussi des « cartes d’importance » au niveau des résidus, aidant les scientifiques à voir quelles parties de chaque peptide motivent les décisions du modèle.
Inventer de nouveaux candidats tout en évitant les fuites de données
Pour compenser le nombre limité de peptides antidiabétiques connus, l’équipe a ajouté une étape de génération qui alimente uniquement le processus d’entraînement. Ils ont utilisé un mélange de stratégies — mutations guidées, recombinaison de motifs et un autoencodeur variationnel — pour proposer de nouvelles séquences qui ressemblent, sans copier, les peptides actifs connus. Ces candidats ont ensuite été filtrés par des « portails de descripteurs » stricts qui imposent des charges, tailles et propensions de liaison réalistes, ainsi que par des outils externes évaluant la similarité avec des peptides bioactifs connus. Seules les séquences passant ces filtres et demeurant clairement distinctes de tous les peptides de test sont conservées comme positifs faiblement étiquetés pour l’entraînement ; aucune n’est jamais utilisée pour évaluer le modèle. Cette approche a élargi l’ensemble d’entraînement tout en préservant un jeu de test propre et non biaisé.
Performance du système et portée des résultats
Lorsqu’on l’a mis au défi avec un panel entièrement indépendant de 180 peptides étudiés expérimentalement et recueillis dans la littérature récente, le cadre a correctement étiqueté environ 99 séquences sur 100, avec précision et rappel proches de 0,99. En termes pratiques, cela signifie qu’il manque rarement un vrai peptide antidiabétique et qu’il classe rarement un peptide inactif comme prometteur. Les analyses des cartes d’attention et des tests de mutation ont montré que le modèle a appris des règles chimiquement sensées : il s’appuie fortement sur des résidus chargés positivement et certains résidus hydrophobes connus pour être importants dans la liaison aux cibles liées au diabète. Des simulations de docking moléculaire ont en outre suggéré que certains des peptides nouvellement générés peuvent établir des contacts plausibles avec le récepteur humain GLP‑1. Si ces prédictions nécessitent encore une confirmation en laboratoire, l’étude démontre une manière reproductible et biologiquement fondée d’explorer l’immense espace des peptides candidats et de prioriser ceux qui ont le plus de chances d’aider à gérer le diabète.
Citation: Asl, Z.R., Rezaee, K., Ansari, M. et al. De novo generation and in silico screening of anti-diabetic peptide candidates via a deep learning–attention framework with physicochemical feature fusion. Sci Rep 16, 6580 (2026). https://doi.org/10.1038/s41598-026-39985-4
Mots-clés: peptides antidiabétiques, apprentissage profond, découverte de médicaments, conception de peptides, récepteur GLP-1