Clear Sky Science · fr
Un apprentissage automatique interprétable explique l’inhibition de l’anhydrase carbonique via la prédiction conforme et contrefactuelle
Pourquoi des médicaments anticancéreux plus intelligents comptent
Les médicaments contre le cancer agissent souvent comme des outils peu précis : s’ils attaquent les cellules tumorales, ils peuvent aussi toucher des tissus sains et provoquer des effets secondaires graves. Une voie prometteuse pour affiner cette action consiste à bloquer des versions spécifiques d’une enzyme appelée anhydrase carbonique, qui aide les tumeurs à survivre en environnement pauvre en oxygène. Cependant, plusieurs isoformes de cette enzyme se ressemblent presque à l’identique, ce qui complique la conception de médicaments qui ciblent les isoformes « nuisibles » dans les tumeurs sans perturber l’isoforme « bénéfique » présente dans tout l’organisme. Cette étude montre comment l’apprentissage automatique interprétable peut aider les chercheurs à relever ce défi et à concevoir des candidats-médicaments plus sélectifs et plus sûrs.
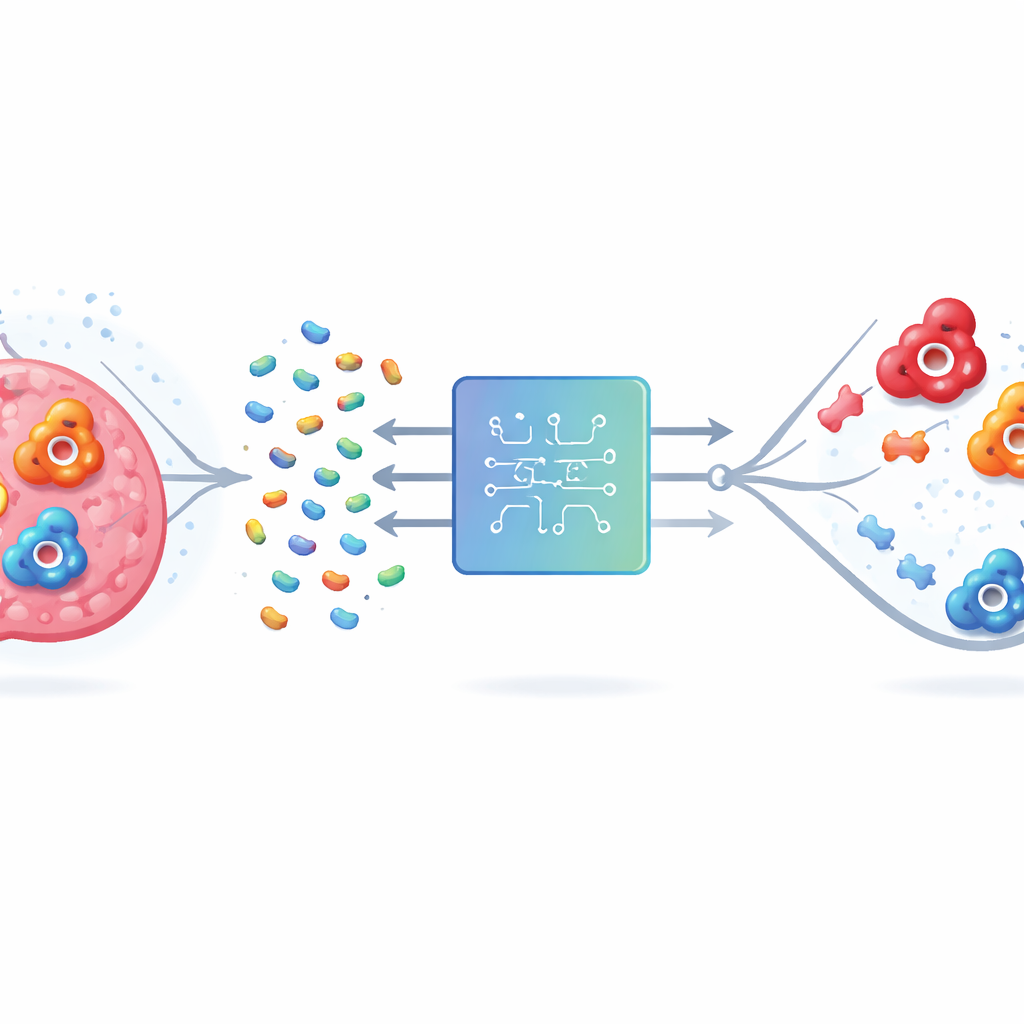
Le problème du mauvais ciblage
L’anhydrase carbonique humaine (hCA) existe sous de nombreuses formes, ou isoformes. Deux d’entre elles, IX et XII, sont liées à la survie des cellules cancéreuses dans les tumeurs appauvries en oxygène, de sorte que leur blocage pourrait ralentir la maladie et améliorer le traitement. Mais l’isoforme II est répandue dans les tissus sains et possède un site actif très similaire à ceux de IX et XII. Les médicaments qui se lient aux trois peuvent déclencher des problèmes indésirables tels que l’acidose métabolique et des troubles de la vision. Les méthodes traditionnelles de laboratoire et de calcul peinent parce que les enzymes sont de grandes molécules complexes et que le nombre de composés potentiellement médicamenteux est astronomique. Les tester exhaustivement, en laboratoire ou en calcul, est tout simplement irréalisable.
Construire une base de données propre et fiable
Les auteurs ont d’abord constitué une base de données soigneusement nettoyée de milliers de molécules testées contre hCA II, IX et XII à partir du dépôt ChEMBL. Ils ont standardisé les structures chimiques, éliminé les mesures douteuses et focalisé sur des composés partageant un groupe de liaison au zinc typique de cette classe d’inhibiteurs. En appliquant des seuils stricts, ils ont étiqueté les molécules comme clairement actives ou clairement inactives et écarté les cas limites qui pourraient perturber les modèles. Comme il y avait beaucoup plus de molécules inactives qu’actives, ils ont équilibré les données pour que les algorithmes d’apprentissage ne favorisent pas simplement la classe majoritaire. Ils ont aussi utilisé une séparation des données basée sur les « échafaudages » moléculaires pour que les ensembles d’entraînement et de test contiennent des cadres moléculaires centraux différents, offrant une image plus réaliste de la capacité des modèles à traiter de véritables composés nouveaux.
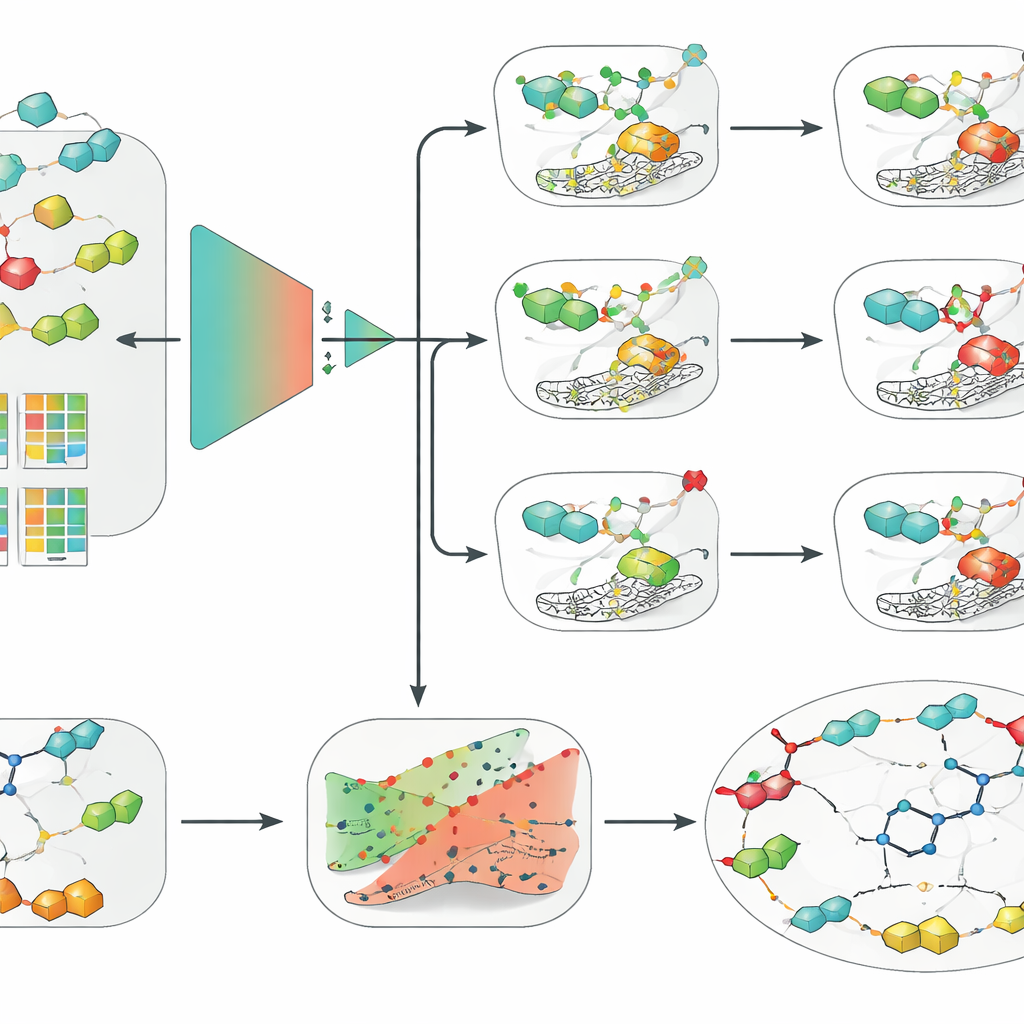
Des modèles simples surpassent le deep learning quand les données sont limitées
Avec cet ensemble de données épuré, l’équipe a comparé un large éventail d’approches, des méthodes classiques d’apprentissage automatique comme la régression logistique, les forêts aléatoires et les machines à vecteurs de support (SVM) aux réseaux neuronaux profonds modernes, incluant des modèles graphiques opérant directement sur les structures moléculaires. Ils les ont combinés avec plusieurs façons d’encoder les molécules : des descripteurs traditionnels faits main, des empreintes clées et des représentations apprises issues d’un modèle de langage chimique. Sur l’ensemble des trois isoformes et sous l’évaluation plus stricte basée sur les échafaudages, une combinaison s’est distinguée de manière consistante : une SVM alimentée par des empreintes à connectivité étendue, une manière structurée de décrire les environnements chimiques locaux au sein d’une molécule. De façon surprenante, cette configuration relativement simple a surpassé des modèles graphiques et profonds plus en vogue, soulignant que la qualité des données, une validation rigoureuse et de bons descripteurs moléculaires peuvent compter davantage que la complexité algorithmique lorsque les jeux de données sont de taille modeste.
Ajouter une confiance fiable et des explications accessibles
Les chercheurs ont ensuite enrichi leur meilleur modèle SVM de deux couches supplémentaires destinées à rendre ses prédictions plus exploitables en découverte de médicaments. Premièrement, ils ont appliqué un cadre appelé prédiction conforme, qui ne fournit pas une réponse binaire unique mais une plage de résultats probables assortie d’un taux d’erreur garanti. Cela permet aux scientifiques de régler le niveau de prudence souhaité et d’identifier les cas où le modèle est véritablement incertain. Deuxièmement, ils ont utilisé des explications contrefactuelles pour rendre le raisonnement du modèle plus intuitif. Pour une molécule donnée, ils ont généré des analogues proches qui inversent le résultat prédit, d’actif à inactif ou inversement. En examinant ces paires pour le candidat clinique SLC-0111, qui bloque sélectivement IX et XII mais pas II, la méthode a redécouvert indépendamment une importante intuition de chimie médicinale : de petits changements dans la partie « queue » de la molécule modifient fortement l’isoforme qu’elle préfère lier.
Des algorithmes aux outils pratiques de conception médicamenteuse
Pour rendre leur approche accessible, les auteurs ont empaqueté les trois modèles SVM, la couche d’incertitude et le moteur contrefactuel dans un outil graphique nommé CAInsight. Un utilisateur peut fournir la représentation textuelle d’une molécule et, en un clic, obtenir l’activité prédite contre hCA II, IX et XII, une estimation de la fiabilité de chaque prédiction et des suggestions de modifications structurelles susceptibles d’augmenter ou diminuer l’activité. Bien que les modèles se concentrent sur la classification actif/inactif plutôt que sur la prédiction en une étape de la puissance exacte ou de la sélectivité, ils reproduisent déjà le comportement connu de candidats réels et distinguent de subtils changements structurels. Les auteurs notent que des jeux de données plus vastes et plus uniformes, ainsi qu’une analyse approfondie du choix des seuils d’activité, pourraient affiner encore les performances.
Ce que cela signifie pour les futurs médicaments anticancéreux
En termes clairs, ce travail montre que des modèles d’apprentissage automatique soigneusement construits et bien expliqués peuvent aider les chimistes à concevoir des médicaments anticancéreux qui mieux distinguent des cibles enzymatiques qui se ressemblent. En combinant des statistiques robustes, des estimations d’incertitude et des exemples intuitifs « et si », le cadre prédit non seulement quelles molécules sont susceptibles de fonctionner, mais suggère aussi pourquoi. Ce type d’intelligence artificielle transparente pourrait accélérer le criblage virtuel, soutenir la conception générative de nouveaux composés et réduire la charge d’essais-erreurs en laboratoire, contribuant in fine à la découverte de traitements plus sélectifs et plus sûrs pour les patients.
Citation: Ghamsary, M.S., Rayka, M. & Naghavi, S.S. Interpretable machine learning rationalizes carbonic anhydrase inhibition via conformal and counterfactual prediction. Sci Rep 16, 8419 (2026). https://doi.org/10.1038/s41598-026-39771-2
Mots-clés: inhibiteurs de l’anhydrase carbonique, apprentissage automatique interprétable, sélectivité des médicaments, prédiction conforme, explications contrefactuelles