Clear Sky Science · fr
Mutations composées hétérozygotes du gène CHAT, une mutation faux‑sens et une variante au site d'épissage, chez deux frères et sœurs atteints du syndrome myasthénique congénital
Quand la respiration faiblit sans prévenir
Certaines nourrissons semblent en bonne santé à la naissance puis cessent soudainement de respirer lors de fièvres bénignes, nécessitant une ventilation en urgence. Pour leurs familles, ces épisodes sont terrifiants et mystérieux. Cette étude examine deux frères et sœurs japonais et relie leurs crises potentiellement mortelles de faiblesse et d'apnée (pauses respiratoires) à de petites altérations d'un seul gène qui aide les nerfs à communiquer avec les muscles. En combinant indices cliniques, séquençage génétique et modélisation protéique assistée par ordinateur, les chercheurs montrent comment ces mutations perturbent probablement une enzyme clé et offrent aux médecins une cible plus claire pour le diagnostic et le traitement.
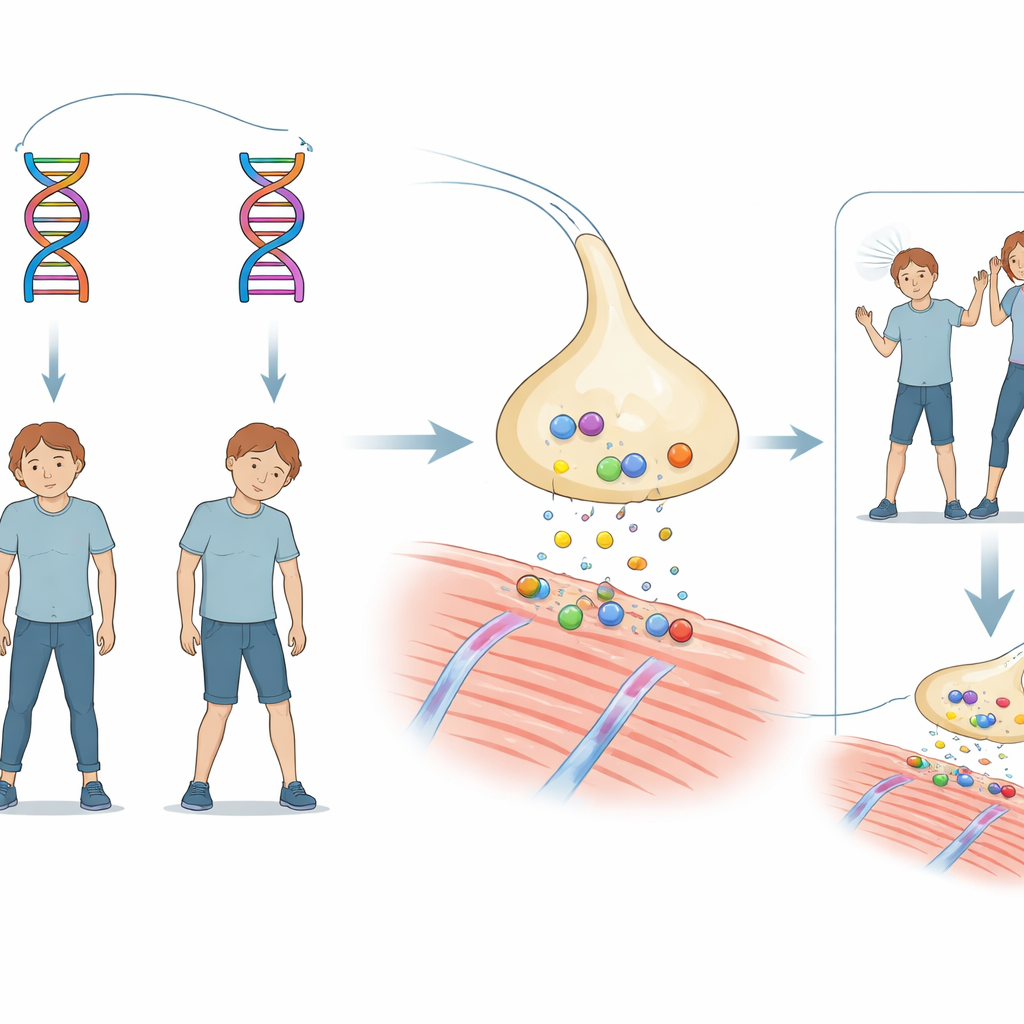
Un mystère familial de faiblesse soudaine
L’histoire concerne un frère et une sœur qui présentaient tous deux un développement moteur légèrement retardé durant la petite enfance. Vers l’âge de 18 mois, chacun a connu des épisodes d’apnée et de perte de connaissance pendant des fièvres, suffisamment graves pour nécessiter une assistance ventilatoire. En grandissant, ils ont tous deux continué d’avoir des épisodes de ptose palpébrale et de faiblesse musculaire générale déclenchés par des infections, la fièvre ou l’effort. L’imagerie cérébrale était normale et les formes communes de myasthénie à médiation auto‑immune ont été écartées. Pourtant, un médicament qui renforce la transmission chimique entre nerfs et muscles a nettement amélioré leurs symptômes, orientant vers une forme héréditaire rare appelée syndrome myasthénique congénital.
Identifier les instructions défectueuses
Pour rechercher une cause héréditaire, l’équipe a séquencé tous les gènes codant des protéines chez les deux enfants et leurs parents. Ils ont trouvé que chaque enfant portait deux altérations différentes dans le même gène, CHAT, qui code pour la choline acétyltransférase — une enzyme qui synthétise l’acétylcholine, le principal messager chimique utilisé par les nerfs pour activer les muscles. Une altération modifiait un seul acide aminé de l’enzyme (une mutation faux‑sens désignée G411R). L’autre se situait à une frontière critique où la cellule coupe et assemble normalement les segments d’ADN lors de la production d’ARN (une mutation du site d’épissage notée c.752+2T>C). Chaque parent portait une seule de ces variations et était en bonne santé ; seuls les enfants ayant hérité des deux présentaient la maladie, ce qui suggère que l’association des deux mutations affaiblit la fonction enzymatique.
Explorer comment une coupure cachée modifie l’enzyme
Comme les chercheurs n’ont pas pu obtenir suffisamment d’ARN CHAT naturel à partir des cellules sanguines, ils ont utilisé une expérience de « minigène ». Ils ont cloné la portion pertinente du gène dans un vecteur d’ADN, introduit la version normale ou la version mutée dans des cellules en culture, puis examiné le traitement de l’ARN. Dans le construct normal, l’ARN contenait tous les segments attendus. Dans la version mutée, un segment entier connu sous le nom d’exon 5 était omis, mais le cadre de lecture global du gène demeurait intact. Cela signifiait que l’enzyme serait produite mais dépourvue d’un court segment interne d’acides aminés. Des comparaisons évolutives ont montré que cette région manquante est fortement conservée entre les espèces, suggérant qu’elle joue un rôle structurel important.
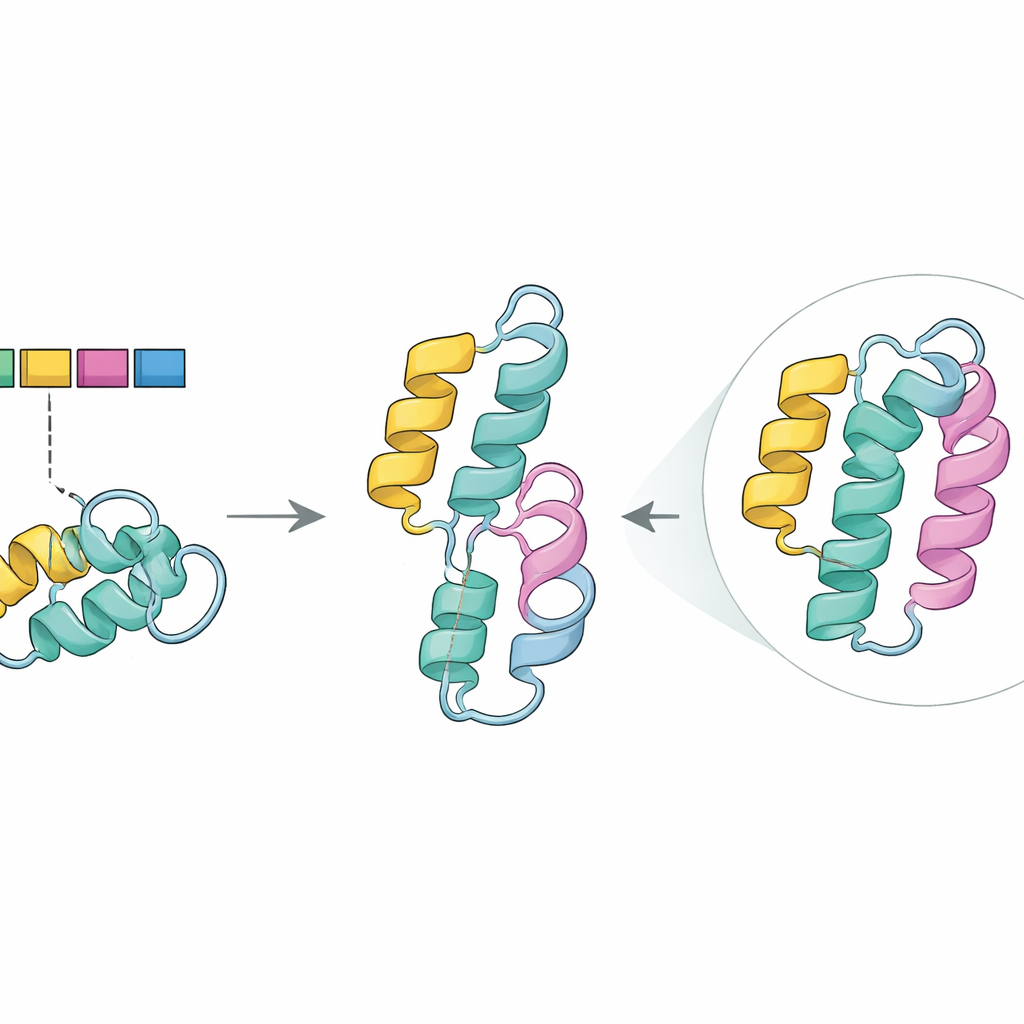
Voir les dégâts structurels in silico
Pour explorer ce rôle, l’équipe s’est tournée vers AlphaFold2, un programme avancé qui prédit la forme tridimensionnelle des protéines à partir de leur séquence. Dans l’enzyme normale, la portion codée par l’exon 5 forme l’un des segments en hélice alpha compactes qui contribuent à stabiliser le cœur de la protéine. Dans la structure prédite pour le mutant, cette hélice avait disparu, laissant un vide dans une région connue pour être cruciale au maintien de la stabilité et au soutien d’une chimie efficace. Associés à des outils informatiques qui identifient les mutations délétères, ces résultats soutiennent l’idée que l’omission de l’exon 5, surtout lorsqu’elle est associée à la substitution G411R sur l’autre copie du gène, compromet la performance de l’enzyme sans l’éliminer complètement — en accord avec les symptômes modérés mais graves observés chez les frères et sœurs.
Implications pour les patients et les familles
L’étude conclut que la combinaison de la mutation faux‑sens G411R et de la nouvelle mutation du site d’épissage dans CHAT est très probablement responsable du syndrome myasthénique congénital chez ces frères et sœurs. En démontrant, par l’essai sur minigène et la modélisation structurelle, comment la variation du site d’épissage supprime une hélice stabilisatrice de l’enzyme, les auteurs fournissent une explication mécanistique sur laquelle cliniciens et chercheurs peuvent s’appuyer. Pour les familles concernées, ce travail apporte plus qu’un nom : il soutient un traitement adapté avec des médicaments qui renforcent la transmission neuromusculaire, oriente le conseil génétique pour les grossesses futures et ajoute un exemple important au catalogue montrant comment de subtiles altérations de notre code génétique peuvent influencer profondément la force musculaire et l’acte fondamental de la respiration.
Citation: Kikuchi, S., Wada, N., Mariya, T. et al. Compound heterozygous CHAT gene mutations, a missense and a splice site variant, in two siblings with congenital myasthenic syndrome. Sci Rep 16, 9346 (2026). https://doi.org/10.1038/s41598-026-39759-y
Mots-clés: syndrome myasthénique congénital, gène CHAT, choline acétyltransférase, mutation du site d'épissage, jonction neuromusculaire