Clear Sky Science · fr
La déconvolution améliorée guidée par attention permet l’estimation des types cellulaires sans référence en transcriptomique spatiale
Voir les cellules in situ
La biologie moderne peut lire l’activité de milliers de gènes simultanément, non seulement dans des cellules isolées mais directement au sein de fines coupes de tissu. Cette vue dite de « transcriptomique spatiale » révèle où vivent et interagissent différents types de cellules, mais chaque mesure mélange souvent les signaux de nombreuses cellules voisines. L’étude présente une nouvelle méthode computationnelle, nommée AGED, capable de démêler ces mixtes et d’estimer quels types cellulaires sont présents où — sans avoir besoin d’un jeu de références de cellules uniques soigneusement apparié. 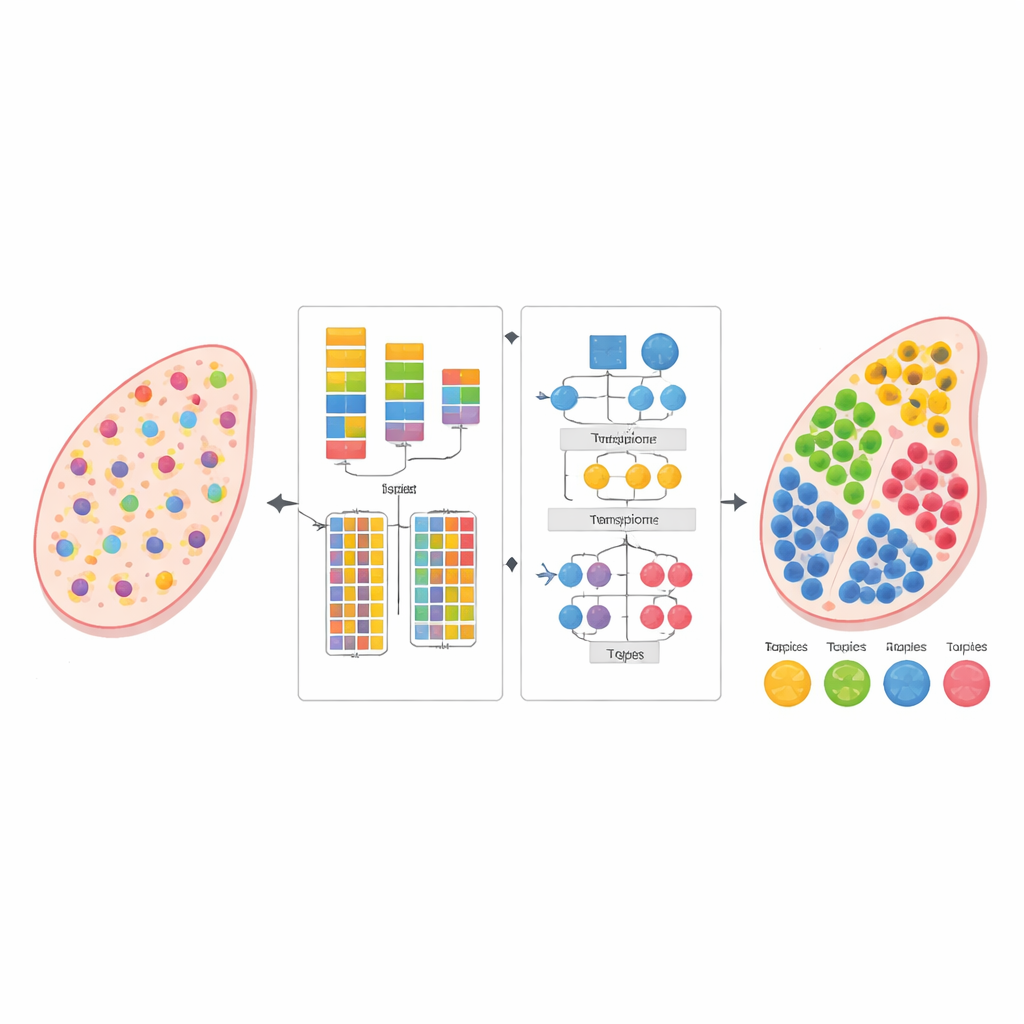
Pourquoi cartographier les cellules dans les tissus est difficile
Les plateformes de transcriptomique spatiale mesurent l’activité génique sur une grille de points appliquée sur une coupe de tissu. Comme la plupart des points capturent plusieurs cellules à la fois, les chercheurs doivent décomposer mathématiquement les signaux mixtes pour retrouver les types cellulaires sous-jacents et leurs proportions. Les outils existants s’appuient souvent sur des atlas de cellules uniques externes du même tissu. Ces atlas peuvent être absents pour des tissus rares, des états pathologiques particuliers ou des conditions expérimentales inhabituelles, et même lorsqu’ils existent ils peuvent ne pas correspondre parfaitement, introduisant des biais. Les méthodes sans référence évitent cette dépendance, mais les approches actuelles peinent face à des motifs spatiaux complexes, des relations géniques subtiles et à la difficulté de déterminer a priori combien de types cellulaires distincts il faut chercher.
Une stratégie en deux étapes pour démêler les mélanges
Les auteurs ont conçu AGED comme un cadre en deux étapes combinant des idées de la statistique et de l’apprentissage profond moderne. Dans la première étape, la méthode teste une gamme de possibilités pour le nombre de types cellulaires susceptibles d’être présents dans le tissu. Elle utilise un réseau neuronal rapide basé sur l’attention, connu sous le nom de Performer, pour apprendre des décompositions candidates puis les évalue selon plusieurs critères simultanément : la qualité de la reconstruction des comptes géniques observés, la séparation nette des groupes cellulaires inférés et la diversité de ces groupes. Une procédure d’ajustement de courbe identifie un « point de coude » où l’ajout de types cellulaires supplémentaires apporte peu d’amélioration, permettant à la méthode de sélectionner automatiquement un nombre approprié plutôt que de se baser sur une estimation de l’utilisateur.
Une attention guidée pour saisir la biologie
Une fois le nombre de types cellulaires déterminé, la seconde étape d’AGED affine la solution avec une architecture d’attention plus riche. Elle part d’un modèle statistique de type « topic model » qui considère chaque point tissulaire comme un mélange de « thématiques » cachées — ici représentant les types cellulaires — et chaque type cellulaire comme un motif génique caractéristique. Ces thématiques initiales fournissent une structure globale. Le modèle superpose ensuite plusieurs mécanismes d’attention : l’un relie les thématiques statistiques au réseau neuronal, un autre agrége l’information des points voisins dans l’espace physique, et un troisième relie directement les thématiques aux gènes. Un système de gating permet au modèle de décider, pour chaque cas, dans quelle mesure il doit faire confiance aux motifs statistiques a priori versus aux données locales. Des contraintes supplémentaires favorisent des solutions parcimonieuses, reflétant la réalité biologique selon laquelle la plupart des emplacements tissulaires sont dominés par seulement quelques types cellulaires principaux. 
Évaluer la méthode
Les chercheurs ont évalué AGED sur plusieurs types de jeux de données. Dans une simulation du bulbe olfactif de souris, la méthode a retrouvé quatre couches anatomiques connues et a rapproché davantage les compositions cellulaires réelles que des outils couramment utilisés avec ou sans référence, atteignant à la fois une forte corrélation avec la vérité terrain et une faible erreur de reconstruction. Dans l’adénocarcinome canalaire pancréatique humain, AGED a automatiquement choisi une solution à vingt types cellulaires qui s’est alignée sur des régions annotées par des pathologistes telles que la tumeur, le canal et le pancréas normal, surpassant d’autres méthodes sur une mesure de similarité structurelle comparant les cartes inférées à la structure tissulaire visible. Dans le thymus humain, AGED a séparé avec précision des populations cellulaires clés et capturé une relation négative biologiquement attendue entre deux types cellulaires épithéliaux spécialisés — un motif que les approches concurrentes n’ont pas réussi à reproduire. Des analyses supplémentaires sur d’autres jeux de données et à une résolution proche de la single-cell ont renforcé la robustesse de la méthode.
Ce que cela implique pour l’avenir
Pour un non-spécialiste, AGED peut être vu comme un moteur intelligent de démélange pour des tissus complexes : il apprend combien de communautés cellulaires distinctes sont présentes, où elles se situent et quels gènes les définissent, le tout à partir des seules données spatiales. En tissant ensemble des modèles statistiques interprétables et des réseaux neuronaux flexibles basés sur l’attention, le cadre offre à la fois précision et compréhension, même en l’absence d’un atlas de référence adéquat. Cela en fait un outil pratique pour explorer l’organisation tissulaire en santé et en maladie, des couches cérébrales aux tumeurs et aux organes immunitaires, et suggère une stratégie plus générale pour utiliser des connaissances a priori afin d’orienter des modèles d’apprentissage automatique puissants mais opaques en biologie.
Citation: Yang, X., Wang, Y. & Chen, X. Attention-guided enhanced deconvolution enables reference-free cell type estimation in spatial transcriptomics. Sci Rep 16, 8097 (2026). https://doi.org/10.1038/s41598-026-39703-0
Mots-clés: transcriptomique spatiale, déconvolution des types cellulaires, apprentissage profond, architecture tissulaire, analyse sans référence