Clear Sky Science · fr
Modèle d’apprentissage analytique piloté par les gènes pour un diagnostic précis du cancer du sein
Pourquoi cette recherche compte pour les patientes et leurs familles
Le cancer du sein est aujourd’hui le cancer le plus fréquemment diagnostiqué chez les femmes dans le monde, et des patientes dont la maladie semble identique sur le papier peuvent avoir des issues très différentes. Cette étude montre comment des motifs dans des milliers de gènes, combinés à un système d’intelligence artificielle soigneusement conçu, peuvent aider les médecins à déterminer de façon plus fiable qui a un cancer et quelle peut en être la sévérité—en n’utilisant que des données réelles de patientes et un ensemble compact de gènes clés.
Des nombreux facteurs de risque au langage des gènes
Le risque de cancer du sein est façonné par de nombreuses influences : mutations héréditaires, hormones, poids corporel, mode de vie, et plus encore. Une fois le cancer apparu, son comportement est déterminé par les gènes qui sont activés ou désactivés à l’intérieur de chaque tumeur. Le séquençage moderne peut mesurer l’activité de dizaines de milliers de gènes simultanément, mais transformer cet océan de chiffres en réponses nettes oui/non pour le diagnostic et le pronostic reste difficile. Les méthodes informatiques traditionnelles étudient souvent les gènes un par un et peuvent manquer la manière dont des groupes de gènes agissent ensemble, ou bien elles performent bien sur un seul jeu de données et échouent lorsqu’on les teste ailleurs.
Apprendre à un modèle à double « cerveau » à lire les motifs géniques
Les auteurs ont construit un modèle d’apprentissage profond « hybride » qui agit un peu comme deux cerveaux spécialisés travaillant de concert. Une partie, inspirée de l’analyse d’images, parcourt une liste ordonnée de gènes pour détecter des motifs locaux—des groupes de gènes dont l’activité conjointe signale un cancer. L’autre partie considère ces mêmes gènes comme une séquence, apprenant comment des gènes « moteurs » précoces et des gènes « aval » plus tardifs s’influencent mutuellement le long de la liste. En combinant ces deux perspectives, le modèle peut saisir à la fois les relations de courte portée et de longue portée au sein de l’empreinte génétique de la tumeur.
Trouver un noyau stable de gènes porteurs de signal
Plutôt que d’alimenter le modèle avec les 17 815 gènes mesurés, l’équipe a conçu un pipeline strict, « sans fuite », pour ne sélectionner que les gènes les plus informatifs. En utilisant une mesure standard de corrélation au sein de boucles de validation croisées répétées, ils ont classé les gènes en fonction de la force avec laquelle leur activité suivait le statut tumoral. Ils ont ensuite conservé uniquement les gènes qui revenaient systématiquement en tête dans tous les plis d’entraînement, aboutissant à une signature stable de 236 gènes. Les chercheurs ont également cartographié les interactions entre ces gènes, montrant que beaucoup forment des réseaux étroitement connectés liés à la croissance tumorale, au métabolisme, à l’immunité et à l’environnement tissulaire—preuve que l’ensemble choisi reflète une biologie réelle et non du bruit aléatoire.
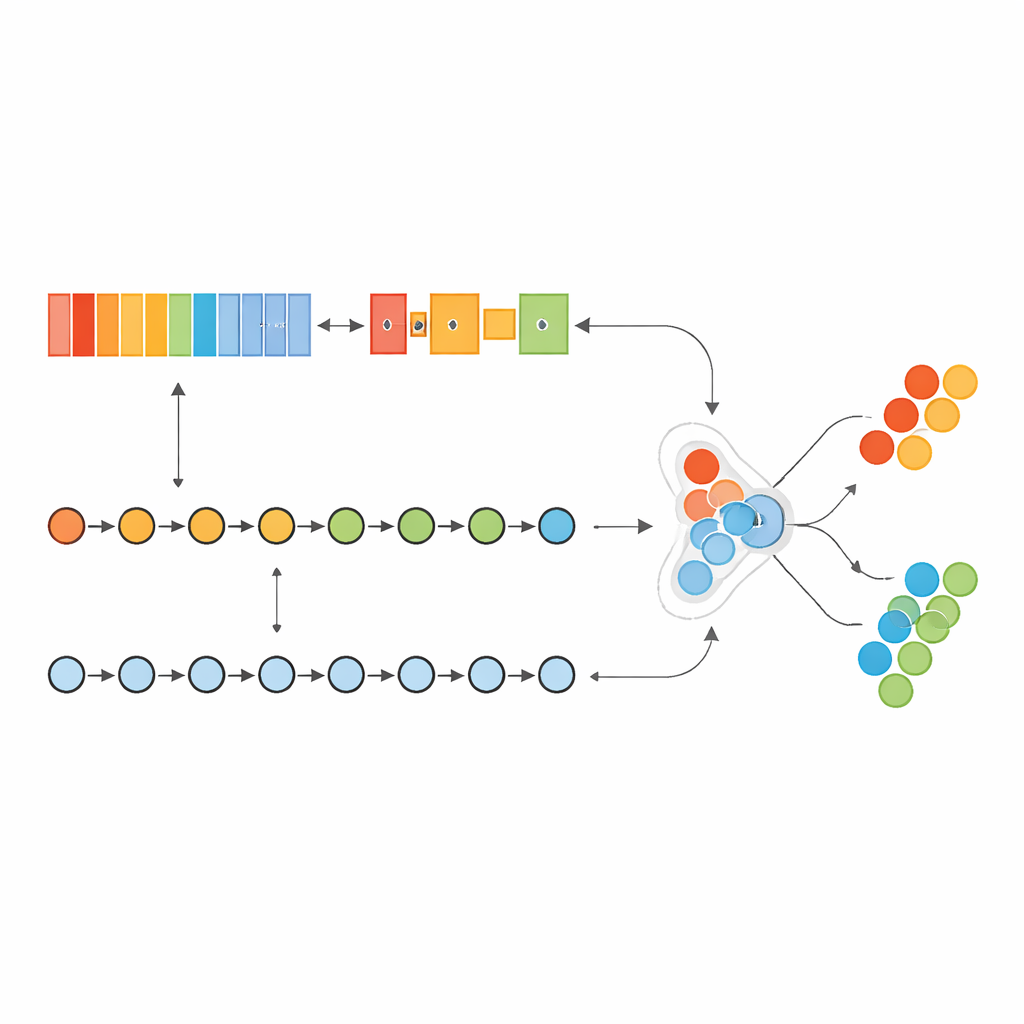
Mettre le modèle à l’épreuve
Le système hybride a été entraîné et ajusté sur des échantillons de cancer du sein issus de The Cancer Genome Atlas puis testé sur un jeu de données entièrement séparé connu sous le nom METABRIC. Pour gérer le fait que les échantillons cancéreux sont bien plus nombreux que les échantillons normaux, les auteurs n’ont pas créé de données artificielles ; ils ont plutôt ajusté l’importance que le modèle accorde aux erreurs sur la classe la plus rare. Après une recherche automatisée des meilleurs paramètres, le modèle a atteint des scores quasi parfaits sur son jeu principal, détectant correctement presque tous les cas de cancer et générant pratiquement aucune fausse alarme. Fait important, la performance est restée extrêmement élevée et très stable même lorsque le modèle a été appliqué à la cohorte externe METABRIC, ce qui suggère que l’approche peut se généraliser au-delà d’une seule étude ou d’un seul hôpital.
Ce que cela signifie pour les soins futurs
En termes simples, ce travail fournit une IA finement optimisée en deux volets qui lit un code compact de 236 gènes pour distinguer les prélèvements mammaires cancéreux des prélèvements non cancéreux avec une précision et une cohérence remarquables, même en conditions bruyantes. Bien que l’étude actuelle ne considère que l’activité génique et utilise des données rétrospectives, ses méthodes posent les bases d’outils futurs pouvant combiner plusieurs types de données—telles que des images tissulaires et d’autres couches moléculaires—et fournir des explications claires sur les gènes qui motivent chaque prédiction. Avec une validation supplémentaire dans des études cliniques prospectives, un tel système pourrait devenir une colonne vertébrale universelle pour le diagnostic de précision du cancer du sein, aidant les médecins à adapter le traitement à la « signature » génétique de la tumeur de chaque patiente.
Citation: Hesham, F., Abbassy, M.M. & Abdalla, M. Gene driven analytical learning model for accurate breast cancer diagnosis. Sci Rep 16, 8155 (2026). https://doi.org/10.1038/s41598-026-39430-6
Mots-clés: diagnostic du cancer du sein, expression génique, apprentissage profond, CNN-BiLSTM, oncologie de précision