Clear Sky Science · fr
CiCLoDS : Regroupement cellulaire conjoint et sélection de gènes pour la transcriptomique spatiale unicellulaire
Trouver des voisinages dans la ville des cellules
Les microscopes modernes peuvent désormais lire quels gènes sont actifs dans des centaines de milliers de cellules tout en conservant chaque cellule à sa place d’origine dans le tissu. Cette révolution de la « transcriptomique spatiale » revient à transformer une carte urbaine floue en une vue au niveau de la rue de chaque maison. Mais il y a un hic : ces cartes contiennent des mesures pour des milliers de gènes par cellule, bien plus que ce que les chercheurs peuvent facilement interpréter ou se permettre de mesurer lors d’expériences de suivi. Cette étude présente CiCLoDS, une nouvelle méthode qui identifie des voisinages cellulaires signifiants et, en même temps, sélectionne une petite liste interprétable de gènes qui définissent ces voisinages.
Une façon plus intelligente de réduire les données massives
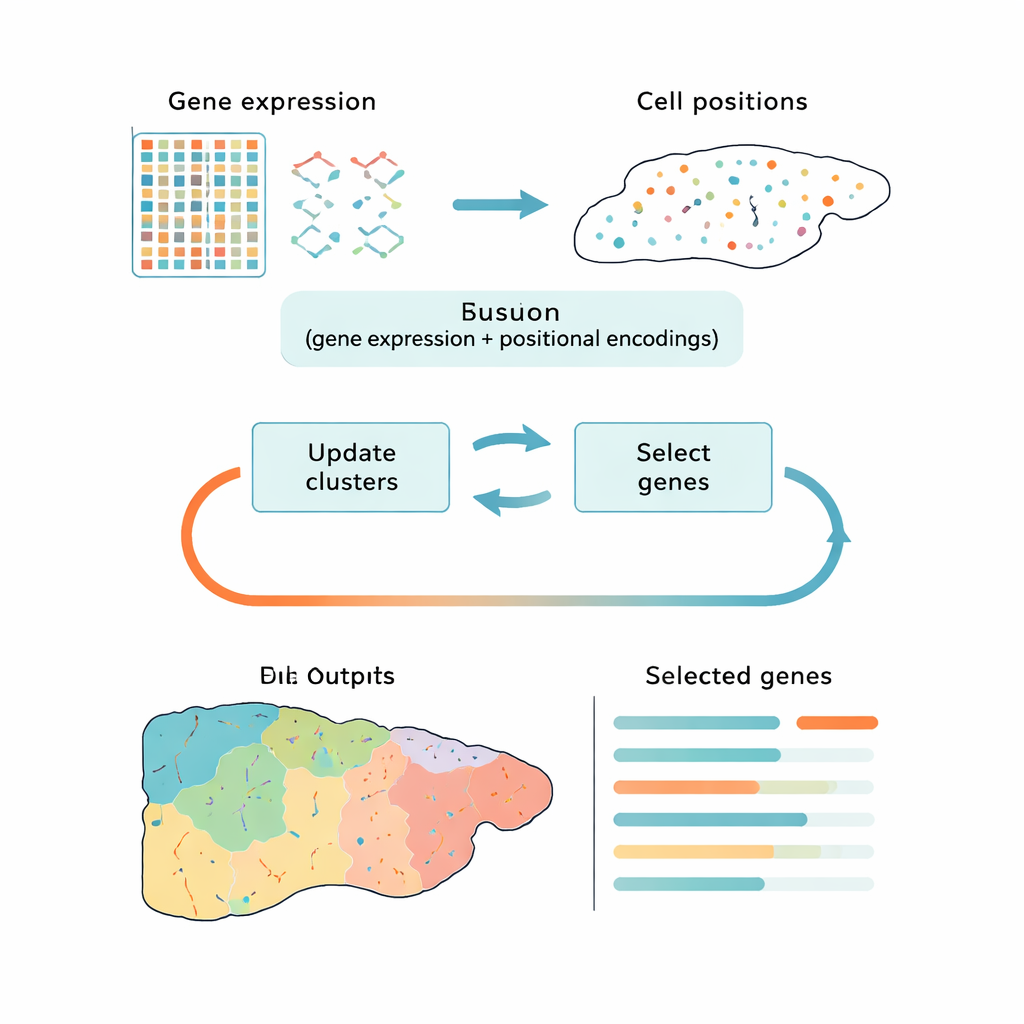
La plupart des outils actuels gèrent ce défi en deux étapes distinctes : d’abord ils réduisent les données à une forme plus simple, puis ils regroupent les cellules en clusters. Des approches populaires comme l’analyse en composantes principales (ACP) préservent la variation globale mais peuvent se focaliser sur des bruits techniques ou des signaux génériques du cycle cellulaire plutôt que sur les différences biologiques pertinentes. D’autres méthodes utilisent l’apprentissage profond pour détecter des motifs, mais elles fonctionnent comme des boîtes noires et n’indiquent pas clairement quels gènes sont les plus importants. CiCLoDS emprunte une voie différente. Il traite la sélection de gènes et le regroupement comme un seul problème conjoint sous une « contrainte budgétaire » définie par l’utilisateur quant au nombre de gènes pouvant être conservés. En pratique, il pose la question suivante : quel ensemble limité de gènes explique le mieux la façon dont les cellules se répartissent en groupes, en tenant compte à la fois de leur activité génique et, lorsqu’elle est disponible, de leur position physique dans le tissu ?
Des mathématiques aux cartes de tissus réels
Les auteurs adaptent une famille de techniques mathématiquement transparentes appelées regroupement en sous‑espaces (subspace clustering) aux réalités de la transcriptomique spatiale, où les jeux de données peuvent contenir plus d’un million de cellules. CiCLoDS fonctionne sur une table simple cellule‑par‑gène, assignant les cellules à des clusters tout en évaluant chaque gène selon sa contribution à la séparation de ces clusters. Il peut aussi intégrer l’information spatiale en ajoutant des « encodages » de position qui décrivent où chaque cellule se situe dans le tissu, sans modifier l’optimisation de base. Sur de grands jeux de données de foie de souris et de côlon humain générés par des plateformes d’imagerie haute résolution, CiCLoDS s’exécute en quelques minutes sur des ordinateurs standards et produit des panneaux géniques compacts — de l’ordre de quelques dizaines à quelques centaines de gènes — qui capturent néanmoins la structure riche des données d’origine.
Révéler des zones cachées et des vaisseaux sanguins
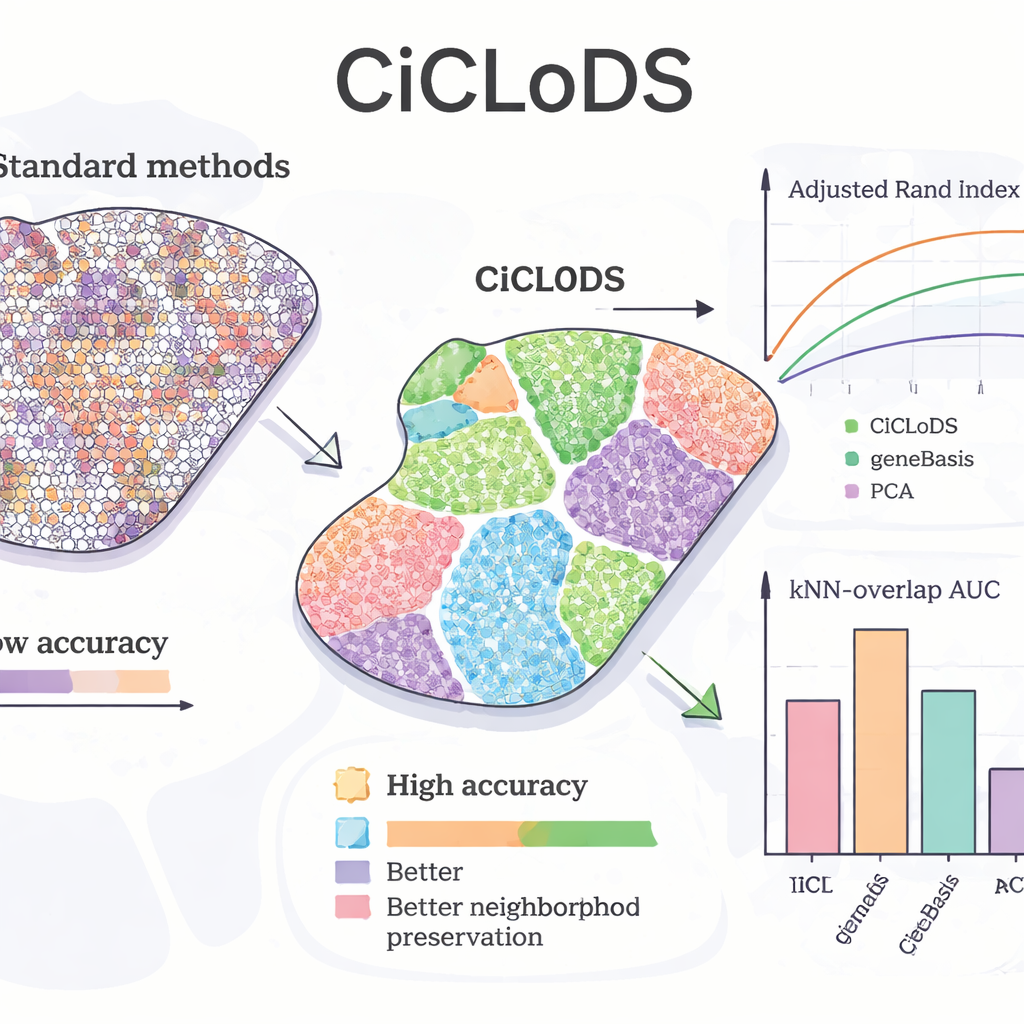
En appliquant CiCLoDS au foie de souris, l’équipe a cherché à savoir si la méthode pouvait retrouver les motifs de « zonation » connus — des changements graduels de la fonction des hépatocytes d’un côté à l’autre du lobule. Par rapport à l’ACP et à un outil de sélection de gènes de référence appelé geneBasis, CiCLoDS a produit des zones spatiales plus nettes avec des frontières plus marquées et beaucoup moins de régions mal assignées, comme le montrent des métriques quantitatives mesurant la concordance avec une carte de référence. Fait remarquable, lorsqu’on lui a permis d’utiliser plus de gènes, CiCLoDS a redécouvert des groupes d’hépatocytes ressemblant à des populations péri‑portal et péri‑centrales qui correspondaient étroitement aux clusters de référence définis par des experts, bien qu’il n’ait pas été informé du biomarqueur clé AXIN2 ni fourni de coordonnées spatiales explicites. Lorsque des encodages spatiaux ont été ajoutés, CiCLoDS a aussi appris des panneaux géniques enrichis pour des fonctions liées aux surfaces cellulaires et aux vaisseaux, et a pu distinguer avec précision de vrais vaisseaux sanguins des artefacts d’imagerie — ce que des méthodes plus simples ont soit manqué, soit n’ont obtenu qu’avec des ajustements ad hoc plus nombreux.
Généraliser aux cerveaux et améliorer d’autres méthodes
Pour vérifier si CiCLoDS tient sur des tissus très différents et entre individus, les auteurs ont analysé des échantillons de cortex préfrontal dorsolatéral humain provenant de trois donneurs. Ici, CiCLoDS a été aussi performant ou meilleur que des méthodes spatiales spécialisées telles que BayesCafe et BayesSpace, en particulier sur un échantillon difficile où les autres outils ont rencontré des difficultés. L’étude met également en avant une utilisation « hybride » : exécuter CiCLoDS d’abord pour obtenir des clusters stables, puis alimenter BayesSpace avec ces clusters. Cette stratégie de démarrage à chaud a amélioré la précision globale et produit des motifs de couches corticales qui correspondaient le mieux aux annotations d’experts, montrant que CiCLoDS peut à la fois fonctionner de façon autonome et rendre les modèles probabilistes en aval plus fiables.
Pourquoi cela compte pour la biologie et la médecine
Pour les non‑spécialistes, l’essentiel est que CiCLoDS transforme des cartes cellulaires écrasantes en résumés concis et biologiquement significatifs. Plutôt que de travailler avec des milliers de mesures bruitées, les chercheurs obtiennent une liste de gènes gérable et des clusters spatiaux clairs qui reflètent l’organisation réelle du tissu — zones métaboliques dans le foie, vaisseaux sanguins et leurs niches, et structures en couches dans le cerveau. Parce que le budget de gènes est contrôlé par l’utilisateur et que les calculs sont légers, CiCLoDS peut aider à concevoir des panneaux géniques ciblés pour des expériences futures, orienter l’interprétation de jeux de données spatiaux complexes et fournir des points de départ robustes pour des modélisations plus élaborées. À une époque où le goulot d’étranglement n’est plus la collecte de données mais leur compréhension, des outils comme CiCLoDS promettent de rendre les cartes tissulaires à haute dimension à la fois pratiques et instructives.
Citation: Wang, N., He, Y., Ray, E. et al. CiCLoDS: Joint cell clustering and gene selection for single-cell spatial transcriptomics. Sci Rep 16, 5356 (2026). https://doi.org/10.1038/s41598-026-39168-1
Mots-clés: transcriptomique spatiale, regroupement cellulaire, sélection de panneaux géniques, architecture tissulaire, analyse unicellulaire