Clear Sky Science · fr
Optimisation computationnelle de la solubilité du domaine calpaïne DEK1 via modélisation structurelle intégrée et mutagenèse ciblée guidée par les données
Pourquoi il importe de rendre les protéines végétales moins récalcitrantes
Beaucoup des protéines qui régulent la croissance des plantes sont de grosses molécules fragiles qui refusent de se dissoudre lorsque les chercheurs tentent de les étudier en laboratoire. L’une d’elles, appelée DEK1, contribue à façonner l’organisme végétal depuis l’échelle cellulaire. Mais comme une portion cruciale de DEK1 s’agglomère quand elle est produite en bactérie, sa structure 3D reste inconnue, ce qui ralentit les efforts pour la comprendre et l’exploiter. Cette étude montre comment la modélisation informatique et une conception intelligente guidée par les données peuvent repenser cette région problématique pour la rendre plus soluble, sans compromettre son architecture — offrant une recette générale pour dompter des protéines difficiles.
Cibler la zone problématique d’une protéine végétale clé
DEK1 est une protéine exceptionnellement grande, ancrée dans les membranes cellulaires et pourvue d’un domaine enzymatique de type calpaïne. Des travaux génétiques ont montré que ce domaine est essentiel au développement normal des plantes comme les mousses et les cultures, pourtant sa structure n’a jamais été résolue expérimentalement. Lorsque les chercheurs tentent d’exprimer ce noyau calpaïne (appelé CysPc) dans la bactérie hôte courante Escherichia coli, il a tendance à devenir insoluble et à former des corps d’inclusion denses. Cela rend quasiment impossible sa purification en quantité et en qualité suffisantes pour des études structurelles et fonctionnelles détaillées. Les auteurs ont donc entrepris de redessiner le domaine CysPc pour qu’il se dissolve plus facilement tout en préservant sa conformation globale.
Construire un modèle 3D fiable à partir de zéro
Faute de structure expérimentale pour cette calpaïne végétale, l’équipe a d’abord dû prédire sa forme 3D. Ils ont combiné plusieurs outils de prédiction de structure de pointe, dont AlphaFold2, SWISS-MODEL et I-TASSER, et ont ancré ces prédictions sur des structures connues de calpaïnes animales apparentées. En adoptant une approche par consensus, ils ont affiné et vérifié les modèles obtenus avec plusieurs tests de qualité évaluant la géométrie de la chaîne principale, le compactage et la conformité aux motifs structurels connus. Ces vérifications indépendantes ont montré que le modèle intégré du domaine CysPc était plus fiable que chaque prédiction prise isolément, fournissant une base solide pour explorer comment de petites modifications de la séquence d’acides aminés pourraient améliorer la solubilité.
Tester des mutations virtuelles dans un solvant simulé
Avec le modèle 3D en main, les auteurs ont réalisé des simulations de dynamique moléculaire étendues, où la protéine et les molécules d’eau environnantes sont suivies au cours du temps sur ordinateur. Ils se sont concentrés sur les résidus en surface de la protéine qui étaient flexibles, hydrophobes ou susceptibles de favoriser l’agrégation. Des positions candidates ont été mutées individuellement en acides aminés plus compatibles avec l’eau puis simulées pendant 200 nanosecondes chacune. Pour chaque variante, ils ont mesuré des caractéristiques liées à la solubilité, comme la surface de contact avec l’eau, la compacité de la protéine et l’amplitude des fluctuations atomiques. De nombreuses mutations simples ont modérément augmenté l’exposition au solvant ou les liaisons hydrogène internes tout en maintenant le repliement global, suggérant que l’ossature de base du CysPc pouvait tolérer des substitutions soigneusement choisies.
Laisser les algorithmes explorer l’espace des mutations
Modifier un seul résidu produit rarement des gains spectaculaires de solubilité, les chercheurs ont donc exploré ensuite des combinaisons de deux et trois mutations. Ils ont généré une bibliothèque de variants doubles et triples construits à partir des meilleures mutations simples et ont de nouveau simulé chacun d’eux. Pour évaluer et classer ces designs de façon équitable, ils ont défini un indice pondéré combinant plusieurs caractéristiques de simulation connues pour corréler avec la solubilité, récompensant l’hydratation et le renforcement des liaisons internes tout en pénalisant une flexibilité excessive. Ils ont ensuite utilisé un algorithme d’apprentissage par renforcement (Proximal Policy Optimization) pour naviguer dans l’immense espace des triples mutants possibles et proposer les combinaisons les plus prometteuses. Cette recherche axée sur les données a convergé vers un mutant triple particulier, nommé MUT347, comme candidat principal.
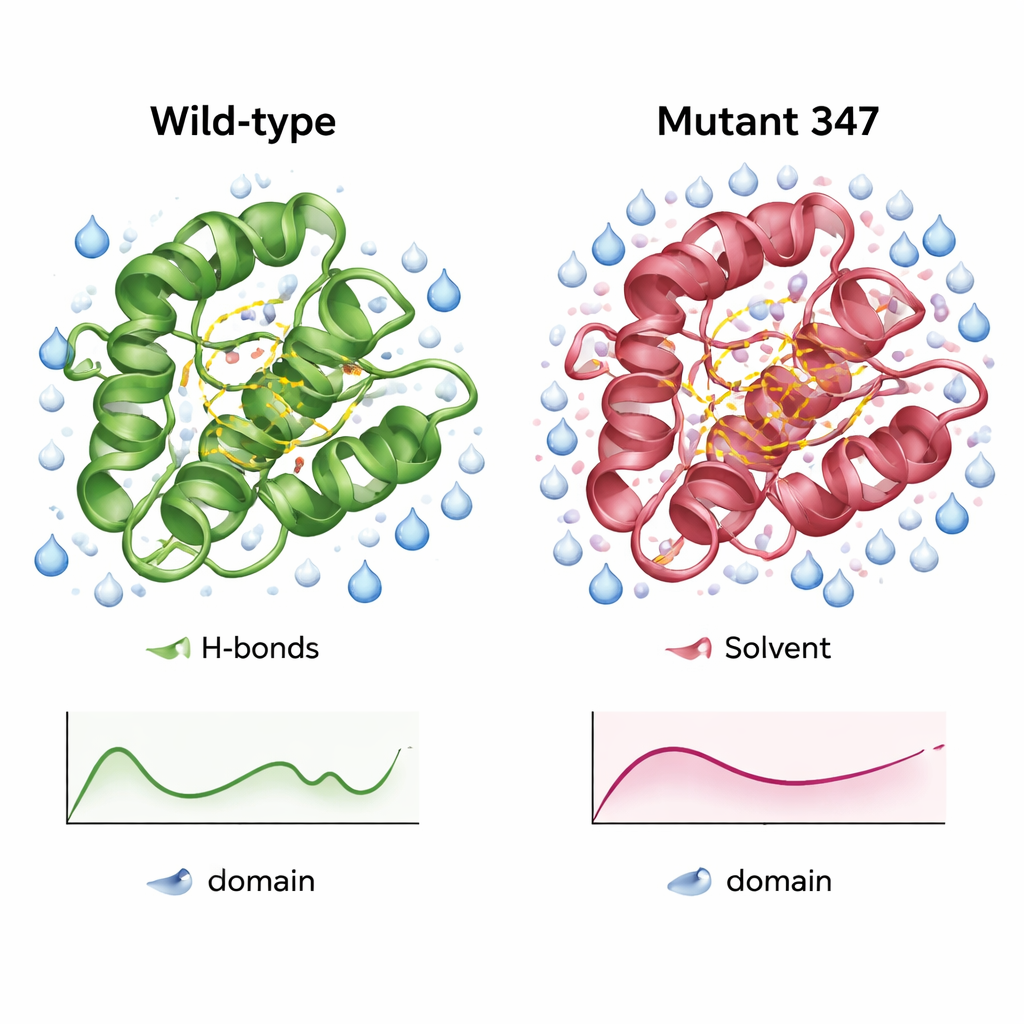
Une version de l’enzyme plus compacte et mieux hydratée
Des simulations détaillées du domaine CysPc sauvage et de MUT347 ont révélé en quoi la variante ingénée différait. MUT347 s’est équilibrée plus rapidement et a montré des écarts globaux plus faibles par rapport à sa forme initiale, indiquant une plus grande stabilité structurelle en solution. Ses boucles et extrémités de chaîne étaient légèrement moins flottantes, tandis que la région catalytique centrale conservait sa flexibilité d’origine, suggérant que les mouvements importants pour la fonction étaient préservés. Le mutant triple présentait davantage de liaisons hydrogène internes et une surface accessible à l’eau plus grande dans des régions clés, signes d’une surface mieux organisée et plus hydratée. Sous différentes concentrations en sel et niveaux de pH, MUT347 a constamment maintenu des fluctuations plus faibles que la protéine d’origine, un comportement associé à une moindre tendance à s’agglomérer.
Ce que cela implique pour l’étude et la réutilisation des protéines
Pour un public non spécialiste, l’essentiel est que les auteurs ont élaboré une recette largement informatique pour transformer un fragment récalcitrant et sujet à l’agrégation d’une protéine végétale essentielle en une version plus soluble et maniable, sans disposer préalablement d’une structure expérimentale. En combinant prédiction moderne de structures, simulations longues et algorithmes d’apprentissage capables de gérer de nombreux choix de conception simultanément, ils ont identifié une mutation triple censée stabiliser le repli et l’exposer plus favorablement à l’eau. Si des travaux expérimentaux restent nécessaires pour confirmer ces améliorations dans des éprouvettes réelles, ce cadre pourrait être utile de manière générale pour sauver d’autres protéines eucaryotes difficiles à produire, aidant en fin de compte les chercheurs à accéder à des structures et fonctions aujourd’hui hors de portée.
Citation: Dabiri, M., Levarski, Z., Struhárňanská, E. et al. Computational optimization of DEK1 calpain domain solubility through integrated structural modelling and data-driven targeted mutagenesis. Sci Rep 16, 7767 (2026). https://doi.org/10.1038/s41598-026-38805-z
Mots-clés: solubilité des protéines, mutagenèse informatique, dynamiques moléculaires, calpaïne végétale DEK1, ingénierie des protéines