Clear Sky Science · fr
Le lncRNA FTX favorise la fibrose myocardique en épuisant miR-335-3p pour réguler la voie TFEC/ILK
Pourquoi la cicatrisation du cœur compte
L’insuffisance cardiaque touche des dizaines de millions de personnes dans le monde et s’installe souvent silencieusement sur plusieurs années. Un moteur majeur de cette détérioration est la fibrose myocardique — la cicatrisation lente et progressive du muscle cardiaque qui le rend plus rigide et moins capable de pomper le sang. Cette étude explore le « câblage » moléculaire qui incite les cellules cardiaques à déposer trop de tissus cicatriciels, et identifie une nouvelle chaîne de molécules susceptibles d’être ciblées pour ralentir, voire inverser, ce processus nocif.
Un examen approfondi de la cicatrisation cardiaque
Lorsque le cœur est blessé ou soumis à un stress, des cellules de soutien appelées fibroblastes cardiaques entrent en action. Dans une réparation saine, elles aident à colmater les lésions. Mais dans les maladies chroniques, elles peuvent basculer vers un état hyperactif, sécrétant un excès de collagène et d’autres composants de la matrice extracellulaire, ce qui finit par raidir la paroi cardiaque. Les chercheurs ont utilisé deux modèles pour étudier ce processus : des souris traitées par isoprotérénol, qui induit de façon fiable une fibrose cardiaque, et des fibroblastes cardiaques humains exposés au facteur de signalisation TGF-β1, un déclencheur bien connu de la cicatrisation. Dans les deux contextes, ils ont mesuré comment des gènes et des protéines spécifiques évoluaient au fur et à mesure du développement de la fibrose.
Une réaction en chaîne délétère à l’intérieur des cellules
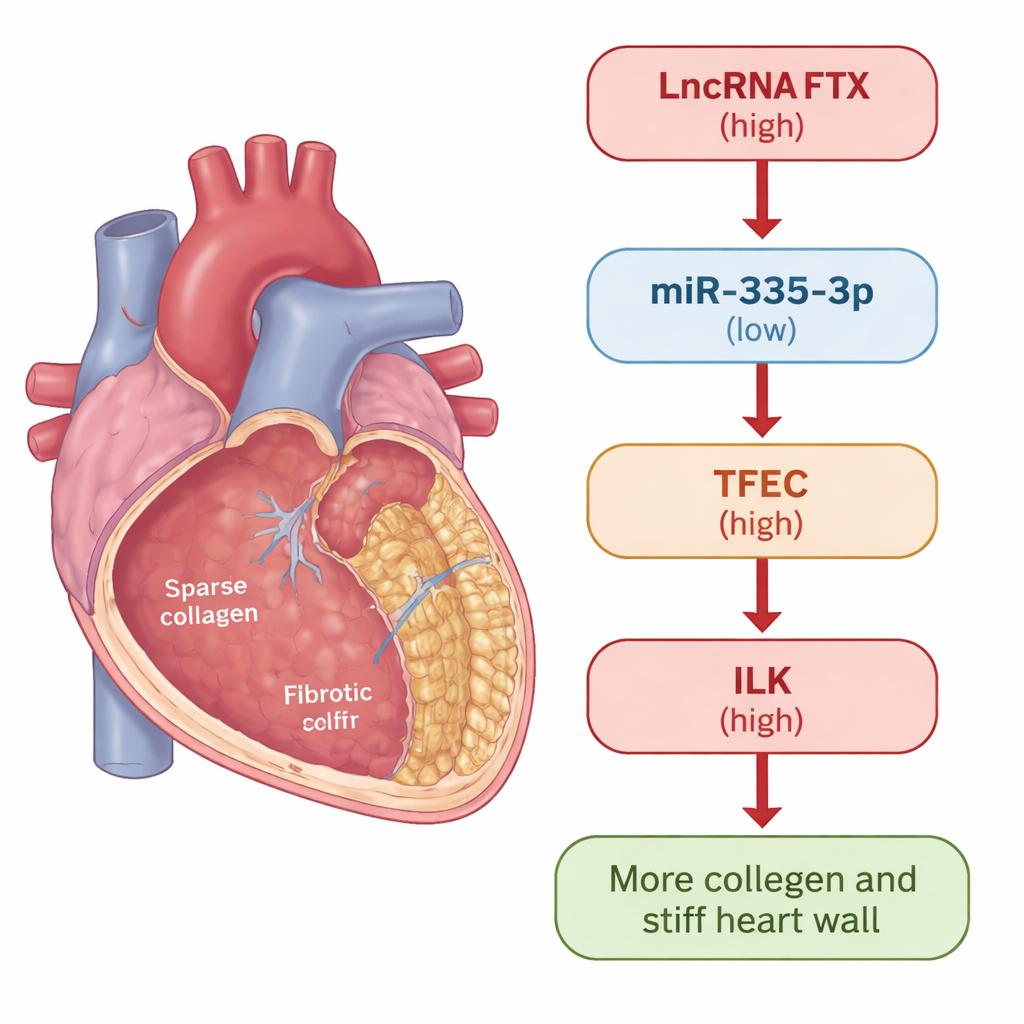
L’équipe s’est concentrée sur un facteur de transcription nommé TFEC, une protéine située dans le noyau cellulaire qui active d’autres gènes. Ils ont constaté que TFEC, ainsi qu’une autre protéine appelée intégrine-linked kinase (ILK), étaient systématiquement augmentés lorsque les fibroblastes basculaient vers un état fibrotique producteur de cicatrices. La réduction de l’expression de TFEC ou d’ILK a fortement diminué les marqueurs classiques de la fibrose tels que l’α-actine musculaire lisse et les collagènes I et III, et a également atténué une voie de contrôle de la croissance (Akt/GSK3β et la signalisation Hippo) connue pour favoriser la cicatrisation tissulaire. Des expériences de cartographie de liaison à l’ADN ont montré que TFEC se fixe directement au promoteur du gène ILK et en augmente l’activité, plaçant clairement TFEC en amont d’ILK dans une cascade de signalisation pro-fibrose.
Des commutateurs ARN qui contrôlent le régulateur maître
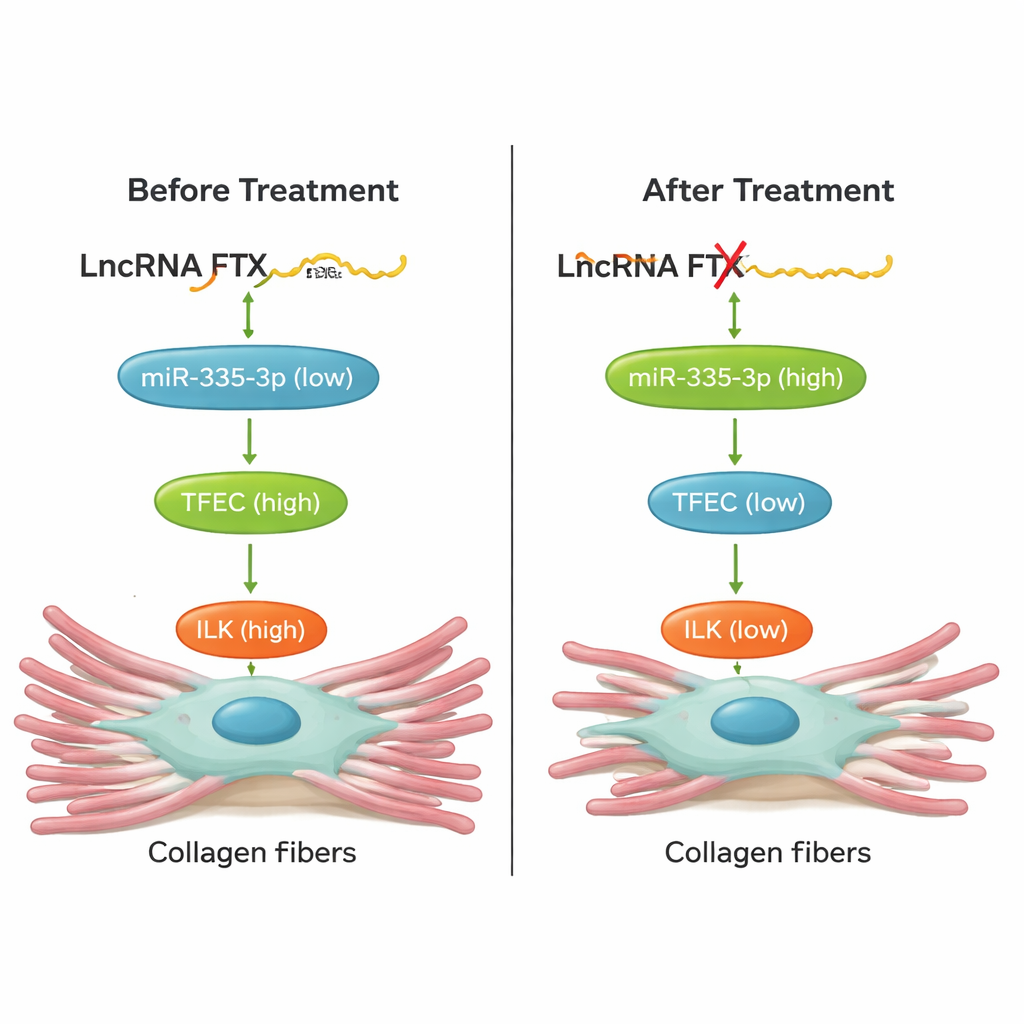
Pour comprendre ce qui contrôle TFEC lui-même, les chercheurs se sont tournés vers les ARN non codants — des molécules d’ARN qui ne codent pas de protéines mais ajustent finement l’activité des gènes. Ils ont identifié un petit ARN, miR‑335‑3p, qui était réduit dans les cœurs et les cellules fibrosés. L’augmentation des niveaux de miR‑335‑3p a abaissé TFEC, tandis que son blocage a élevé TFEC, et des essais rapporteurs ont confirmé que miR‑335‑3p se lie directement aux messages de TFEC pour en limiter l’expression. Ils ont ensuite identifié un long ARN non codant, appelé FTX, qui était augmenté lors de la fibrose et interagissait physiquement avec miR‑335‑3p. FTX agissait comme une éponge moléculaire : il absorbait miR‑335‑3p, empêchant ce petit ARN de retenir TFEC. En conséquence, TFEC et ILK augmentaient, et les fibroblastes produisaient davantage de collagène formant la cicatrice.
De la culture cellulaire aux cœurs vivants
Surtout, l’équipe a testé si perturber cette chaîne pouvait réellement protéger les cœurs chez l’animal. Chez des souris exposées à l’isoprotérénol, la diminution de TFEC, l’inhibition de FTX dans le cœur via un vecteur de thérapie génique AAV9, ou l’augmentation de miR‑335‑3p avec un « agomir » chimiquement stabilisé ont tous entraîné moins d’accumulation de collagène et des niveaux réduits de marqueurs de fibrose dans le tissu cardiaque. Ces interventions ont également amélioré la fonction cardiaque : le volume d’éjection et la fraction d’éjection se sont rapprochés de la normale, et les augmentations néfastes de la fréquence cardiaque ont été atténuées. Des expériences de sauvetage en culture cellulaire ont montré que modifier un composant de l’axe FTX/miR‑335‑3p/TFEC/ILK faisait évoluer de manière prévisible les autres composants, confirmant qu’il s’agit d’une voie étroitement connectée plutôt que d’une simple corrélation lâche.
Ce que cela signifie pour les traitements futurs
Pour un non-spécialiste, l’idée principale est que les auteurs ont identifié un nouveau « levier de contrôle » de la cicatrisation cardiaque. Un long ARN appelé FTX lève le frein (miR‑335‑3p) sur un commutateur maître (TFEC), qui active ensuite ILK et des signaux pro-cicatrisation en aval, entraînant un dépôt excessif de collagène et une rigidification du cœur. En réduisant FTX, en restaurant miR‑335‑3p ou en bloquant directement TFEC, il a été possible chez la souris de diminuer la fibrose et d’améliorer la fonction de pompage. Bien que des travaux supplémentaires soient nécessaires pour confirmer cette voie chez l’humain et développer des thérapies sûres, cette chaîne de régulation basée sur des ARN offre plusieurs points d’intervention prometteurs pour lutter contre l’insuffisance cardiaque d’origine fibrotique.
Citation: Yao, F., He, Z., Zheng, C. et al. LncRNA FTX promotes myocardial fibrosis by sponging miR-335-3p to regulate TFEC/ILK signaling. Sci Rep 16, 7340 (2026). https://doi.org/10.1038/s41598-026-38615-3
Mots-clés: fibrose myocardique, insuffisance cardiaque, ARN non codant, fibroblastes cardiaques, signalisation de la fibrose