Clear Sky Science · fr
Approches intégrées pour explorer les changements spatio‑temporels du réassortiment génétique du virus influenza aviaire hautement pathogène A(H5) en Eurasie, 2000–2023
Pourquoi les « zones de remix » génétiques de la grippe aviaire nous concernent
La grippe aviaire n’est plus seulement un problème pour les poules et les canards de fermes lointaines. Une forme hautement dangereuse d’influenza aviaire, connue sous le nom de H5, se propage en Europe et en Asie depuis plus de vingt ans, tuant des oiseaux sauvages, décimant des cheptels avicoles et infectant parfois des mammifères, y compris des bovins et des humains. Cette étude pose une question simple mais urgente : où et dans quelles conditions le virus est‑il le plus susceptible de « remixer » ses gènes pour produire de nouveaux variants potentiellement plus dangereux — et comment repérer ces zones à risque à l’avance ?
Suivre un virus qui change de forme
Les virus influenza portent leur matériel génétique en huit segments séparés, qui peuvent être échangés lorsque deux souches différentes infectent le même oiseau. Ce processus, appelé réassortiment, peut créer des combinaisons virales entièrement nouvelles. Les chercheurs ont rassemblé plus de 300 000 séquences génétiques d’influenza issues de bases de données mondiales et, à l’aide d’un pipeline standardisé, les ont regroupées en familles génétiques pour chacun des huit segments. Ils ont ensuite défini 136 « génotypes » génétiques distincts des virus H5 hautement pathogènes circulant dans le monde entre 1996 et 2023. En retraçant où et quand ces génotypes sont apparus, ils ont pu reconstruire le paysage changeant des virus H5 au fil du temps.
Trois vagues de changement viral
L’équipe a constaté que l’évolution du H5 en Eurasie s’est déroulée en trois grandes vagues. De 2000 à 2013, un génotype principal a dominé les foyers, principalement en Asie et dans certaines parties de l’Afrique, provoquant des épisodes sporadiques mais graves dans les élevages. Vers 2014, une nouvelle branche du H5, connue sous le nom de clade 2.3.4.4, a émergé et a inauguré une deuxième vague. Pendant la période 2014–2021, de nombreux génotypes différents ont coexisté et se sont propagés tant chez les oiseaux sauvages que dans les cheptels domestiques, notamment en Europe, en Asie et plus tard dans les Amériques. Une troisième vague a commencé vers 2021 avec l’essor du clade 2.3.4.4b H5N1, qui s’est installé dans plusieurs régions et a provoqué des foyers permanents — un schéma « endémique » plutôt que des pics hivernaux occasionnels. 
Cartographier des points chauds cachés
Pour identifier où les échanges génétiques étaient les plus intenses, les scientifiques ont divisé l’Eurasie en carrés de 100 kilomètres et ont compté combien de génotypes H5 différents y étaient détectés. En utilisant une statistique spatiale mettant en évidence les regroupements, ils ont repéré des « hotspots » de réassortiment — des zones où de nombreux génotypes cooccurrents étaient plus fréquents qu’attendu. Au début, ces hotspots se concentraient en Asie du Sud‑Est. Lors de la deuxième vague, ils se sont déplacés vers le nord et l’ouest, apparaissant le long des côtes pacifiques de l’Asie de l’Est et à travers l’Europe centrale et occidentale, y compris des régions du Danemark, du sud de la Suède et du nord de l’Italie. Ces motifs suggèrent que la géographie et les pratiques agricoles ont toutes deux orienté l’évolution du virus.
Communautés d’oiseaux, élevages et environnement
Les hotspots ne résultent pas d’une seule espèce d’oiseau « problématique » ou d’un seul type d’exploitation ; ils se développent là où de nombreux facteurs se chevauchent. L’équipe a combiné les observations d’oiseaux issues de la science participative via le projet eBird avec des cartes d’occupation du sol, des données de densité avicole et des registres de foyers H5 en élevage. Ils ont d’abord identifié les espèces d’oiseaux sauvages qui avaient tendance à être présentes dans les grilles hotspot, en se concentrant sur trois grands ordres : les anatidés tels que canards et oies (Anseriformes), les limicoles et autres oiseaux côtiers (Charadriiformes), et les passereaux (Passeriformes). Fait surprenant, de nombreuses espèces à haut risque n’avaient jamais été formellement testées pour la grippe aviaire. Pour capturer l’effet combiné de plusieurs espèces, les auteurs ont construit un « score de risque polyspecifique » résumant la probabilité qu’une communauté d’oiseaux locale favorise le réassortiment. Ils ont ensuite ajouté des informations sur les densités de poulets et de canards, les foyers en élevage, et les types de terres tels que les zones cultivées ou urbanisées pour estimer quelles combinaisons de conditions prédisaient le mieux les hotspots.
Des zones humides aux poulaillers
L’analyse a révélé un glissement de la niche écologique du virus. Aux premières années, le réassortiment était principalement associé à l’élevage de canards, cohérent avec le rôle des canards comme réservoir discret portant le virus sans signes clairs de maladie. Au fil du temps, alors que les virus H5 hautement pathogènes se sont installés dans les élevages de poulets — favorisés dans certaines régions par une circulation prolongée et des pratiques de vaccination —, les signaux les plus forts se sont déplacés vers les zones à forte densité de poulets et les paysages agricoles mixtes. Les zones urbanisées dans certaines parties de l’Asie et les terres cultivées en Europe ont également été corrélées aux hotspots, reflétant probablement les lieux où humains, élevages et oiseaux sauvages se rencontrent. Parallèlement, des oiseaux non‑anatidés tels que les passereaux, qui vivent en grand nombre autour des champs, des banlieues et des granges, semblaient de plus en plus jouer un rôle de passerelle entre habitats sauvages et poulaillers. 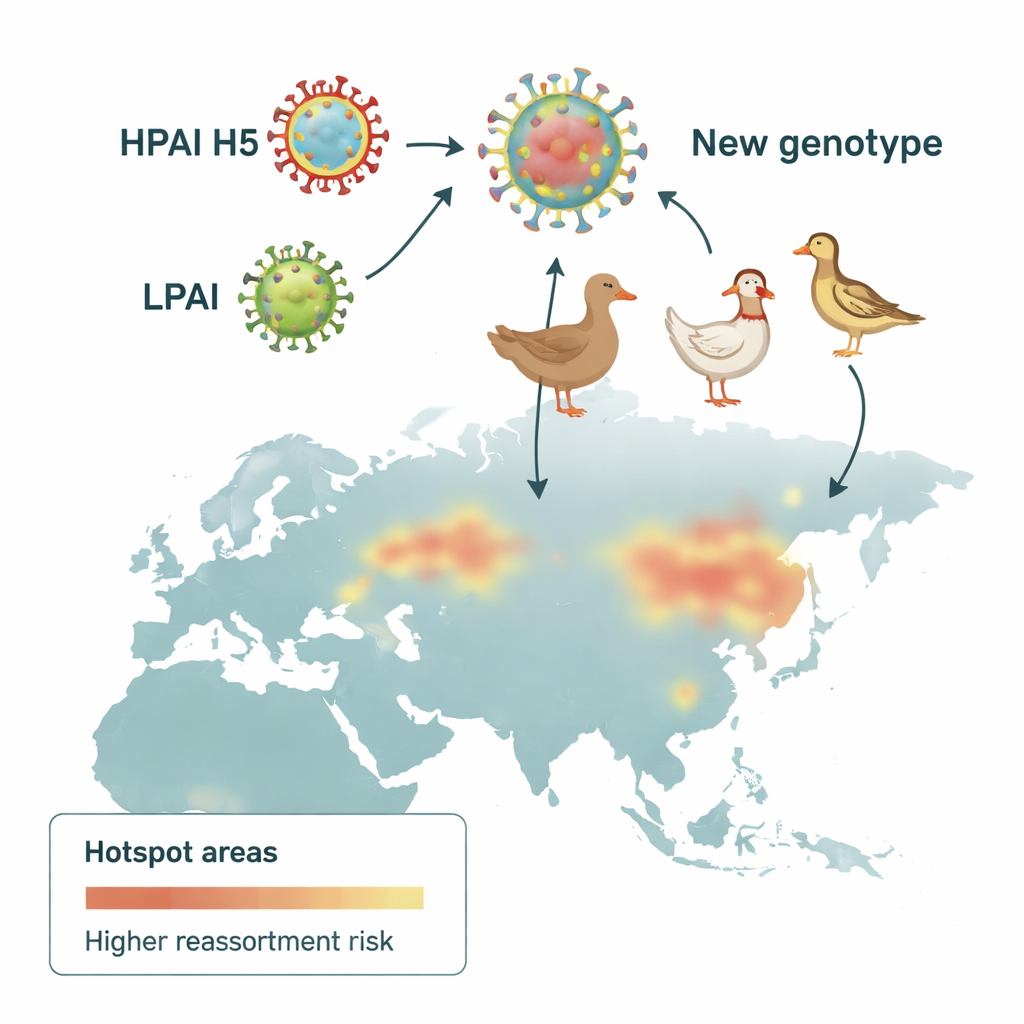
Ce que cela implique pour la préparation
Pour les non‑spécialistes, le message essentiel est que de nouvelles formes dangereuses du virus H5 ont le plus de chances d’apparaître là où l’élevage avicole dense, des communautés d’oiseaux sauvages diversifiées et des paysages modifiés par l’homme se rencontrent. En fusionnant données génétiques, observations ornithologiques et informations environnementales en cartes de risque unifiées, cette étude fournit un guide pour cibler au mieux la surveillance — qu’il s’agisse de tester des groupes d’oiseaux peu étudiés, de renforcer la biosécurité autour des élevages à haut risque, ou de surveiller les régions où le virus est devenu endémique. Comprendre et surveiller ces « zones de remix » génétiques est une démarche pragmatique pour réduire la probabilité qu’un virus animal nous surprenne par un nouveau saut de l’aire de répartition, de la virulence ou des espèces hôtes.
Citation: Chen, BJ., Liang, CC., Li, YT. et al. Integrated approaches to explore temporal-spatial changes in gene reassortment of highly pathogenic avian influenza A(H5) virus in Eurasia, 2000–2023. Sci Rep 16, 7518 (2026). https://doi.org/10.1038/s41598-026-38466-y
Mots-clés: grippe aviaire, H5N1, oiseaux sauvages, élevage avicole, évolution virale