Clear Sky Science · fr
Potentiel interatomique par apprentissage automatique pour les propriétés structurelles des oxydes de fer
Pourquoi les roches rouillées comptent
Les oxydes de fer – les minéraux qui donnent la couleur à la rouille – soutiennent discrètement une grande partie de la vie moderne. Ils constituent la principale source de fer pour l’acier, des ingrédients clés des batteries et des cellules solaires, et contribuent même à l’assainissement des eaux polluées. Pourtant, malgré leur importance, il reste difficile de prédire le comportement de ces matériaux dans des conditions réelles, en particulier au niveau atomique. Cet article décrit comment des chercheurs ont utilisé l’intelligence artificielle moderne pour construire un modèle numérique rapide et précis d’un oxyde de fer crucial, l’hématite, ouvrant la voie à des expériences virtuelles plus fiables, de l’extraction du minerai aux dispositifs d’énergie propre.
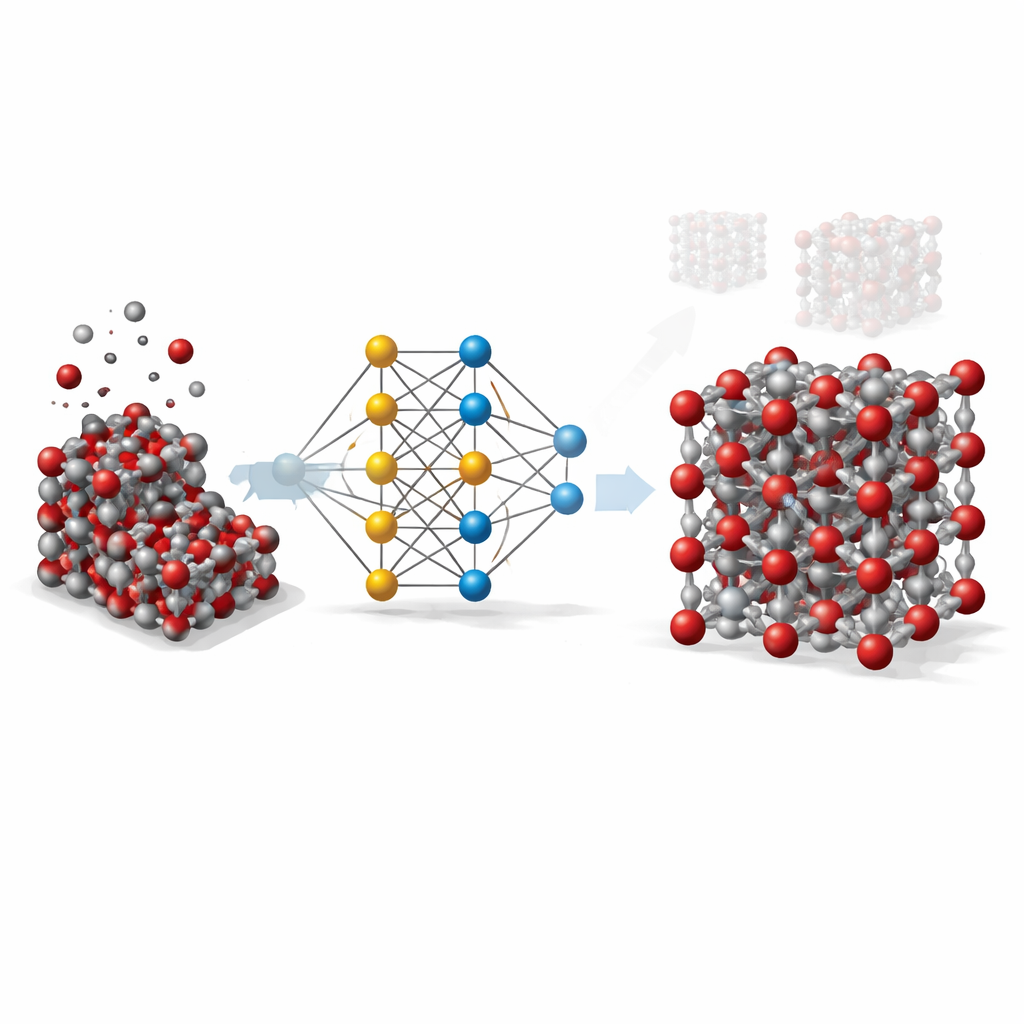
Des calculs coûteux aux raccourcis intelligents
Pour comprendre un solide comme l’hématite en détail, les scientifiques s’appuient idéalement sur des méthodes quantiques qui suivent l’interaction des électrons et des atomes. Ces méthodes, bien que très précises, sont tellement coûteuses en calcul qu’elles deviennent impraticables pour simuler de grands échantillons ou de longues échelles temporelles. Les modèles classiques, en revanche, sont rapides mais approximatifs : ils reposent sur des formules simples ajustées à des situations particulières et échouent souvent lorsque la température, la pression ou la forme du cristal changent. Le travail présenté ici vise à combler cet écart en utilisant l’apprentissage automatique pour imiter la précision des calculs quantiques tout en conservant la rapidité des modèles traditionnels.
Apprendre à un réseau neuronal le comportement des atomes
L’équipe a construit ce qu’on appelle un potentiel basé sur un réseau neuronal graphique pour l’hématite. Dans cette approche, chaque atome est traité comme un nœud du réseau, et les liaisons et atomes voisins forment les connexions entre nœuds. Pour apprendre au réseau comment les atomes de l’hématite s’attirent et se repoussent, les chercheurs ont d’abord généré des milliers d’instantanés atomiques à l’aide de simulations classiques couvrant une large gamme de températures, de pressions et de distorsions cristallines, incluant à la fois des cristaux en volume et des surfaces exposées. Ils ont ensuite utilisé une méthode quantique de haut niveau (DFT+U) pour calculer l’énergie, les forces et les contraintes internes de chaque instantané, et entraîné le réseau neuronal à reproduire ces valeurs aussi fidèlement que possible.
Vérifier le modèle par rapport à la réalité
Une fois entraîné, le nouveau potentiel – appelé Fe-MLIP – a été testé de manière rigoureuse. Les auteurs ont comparé ses prédictions pour des grandeurs structurelles de base, telles que les dimensions du réseau et la déformation du cristal sous contrainte, avec des mesures expérimentales et plusieurs modèles classiques largement utilisés. Fe-MLIP a reproduit la structure cristalline connue de l’hématite à quelques pourcents près et a capturé son comportement élastique presque aussi bien que les calculs quantiques directs, surpassant nettement d’autres champs de force pour de nombreuses propriétés. Il a aussi bien performé sur des tests plus subtils, comme l’expansion thermique du matériau et les vibrations atomiques, importantes pour le transport de la chaleur et la spectroscopie. Ces fréquences vibratoires, qui n’avaient jamais été fournies explicitement pendant l’entraînement, étaient plus proches des valeurs mesurées que celles obtenues avec les modèles concurrents.
Aller au-delà d’un seul minéral
Les chercheurs ont ensuite exploré jusqu’où ce modèle basé sur l’hématite pouvait être étendu. Ils l’ont appliqué à des oxydes de fer apparentés – la maghémite et la magnétite – qui partagent des blocs atomiques similaires mais diffèrent par l’arrangement cristallin et les états de charge du fer. Même si Fe-MLIP n’avait pas été entraîné sur ces phases, il a produit des valeurs raisonnables pour leurs paramètres de réseau et leur rigidité, égalant souvent voire surpassant des modèles classiques spécialisés. Le potentiel a également capturé la stabilité relative des surfaces cristallines clés et même les tendances du coût énergétique de création de lacunes atomiques, des caractéristiques cruciales pour comprendre la corrosion, la catalyse et les performances des batteries.
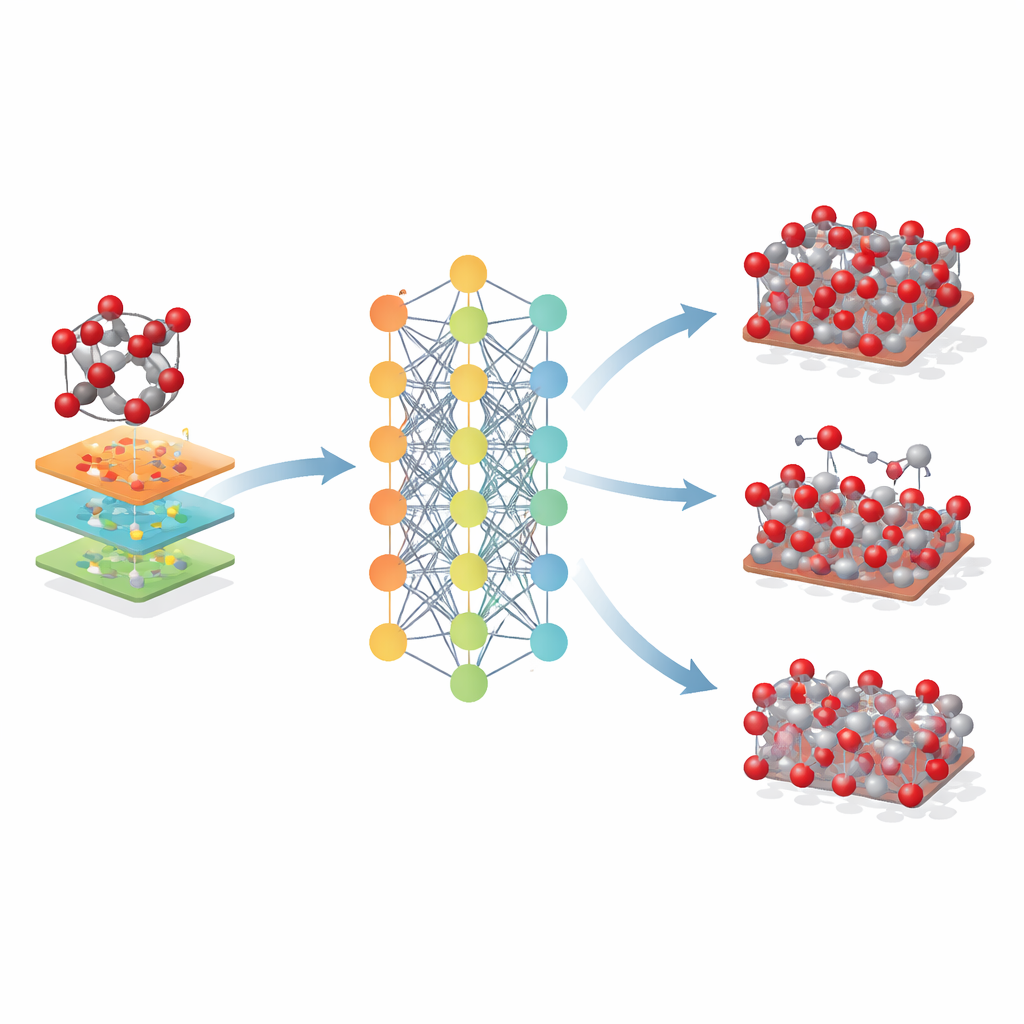
Ce que cela signifie pour la conception future des matériaux
Pour les non-spécialistes, la conclusion est que ce travail fournit un nouveau « jumeau numérique » puissant pour les oxydes de fer. Le modèle Fe-MLIP permet aux chercheurs de réaliser de grandes simulations longues de l’hématite et des matériaux apparentés avec une fiabilité proche du niveau quantique mais à une fraction du coût. Bien qu’il hérite de certaines limites de la méthode quantique sous-jacente et soit actuellement centré sur le fer et l’oxygène, il permet déjà des études plus réalistes de la réponse de ces minéraux à la contrainte, à la chaleur, aux surfaces et aux défauts. En termes pratiques, un tel outil peut accélérer la conception de procédés sidérurgiques améliorés, de catalyseurs et de batteries plus efficaces, et d’outils environnementaux optimisés reposant sur les oxydes de fer – le tout en permettant aux scientifiques de tester des idées sur ordinateur avant de passer au laboratoire ou à la mine.
Citation: Torres, A., de Oliveira, A.B., Barbosa, M.d.S. et al. Machine learning interatomic potential for the structural properties of iron oxides. Sci Rep 16, 8576 (2026). https://doi.org/10.1038/s41598-026-38096-4
Mots-clés: hématite, oxydes de fer, potentiel par apprentissage automatique, réseaux neuronaux graphiques, dynamique moléculaire