Clear Sky Science · fr
Caractérisation moléculaire des troubles ataxiques et neuropathiques à transmission récessive dans des familles pakistanaises consanguines
Pourquoi c’est important pour les familles et les médecins
Les troubles de l’équilibre, de la marche et de la sensibilité des mains et des pieds peuvent être profondément invalidants, surtout lorsqu’ils débutent pendant l’enfance et s’aggravent lentement tout au long de la vie. Pour de nombreuses familles, en particulier dans les régions où les mariages entre cousins sont fréquents, ces symptômes se transmettent sur plusieurs générations sans explication claire. Cette étude aborde une question cruciale pour de telles familles au Pakistan : les analyses d’ADN modernes peuvent‑elles enfin révéler les causes génétiques cachées de leur ataxie (troubles de l’équilibre et de la coordination) et de leur neuropathie périphérique (atteinte des nerfs des membres), et aider les cliniciens à proposer des diagnostics plus précis et des pistes de traitement ?
Suivre la maladie héréditaire au sein de grandes familles
Les chercheurs ont travaillé avec sept familles pakistanaises étendues dont plusieurs membres souffraient de troubles moteurs et nerveux sévères. Certaines personnes présentaient essentiellement une ataxie, rendant la marche instable et altérant le contrôle de la parole et des mouvements oculaires. D’autres montraient des signes classiques de neuropathie périphérique, comme l’atrophie musculaire des mains et des pieds, des déformations plantaires et une perte des réflexes. Dans ces familles, les parents étaient apparentés, ce qui augmente la probabilité que les enfants héritent de deux copies d’un même gène rare délétère. À partir d’échantillons sanguins de proches affectés et non affectés, l’équipe a réalisé un séquençage de l’exome — lecture de presque toutes les régions codant pour des protéines du génome — pour rechercher des variants délétères se transmettant avec la maladie dans les arbres généalogiques. 
Localiser des gènes rares délétères
En filtrant les variantes d’ADN communes et bénignes, les scientifiques ont identifié des variants probablement responsables de la maladie dans cinq des sept familles. Chacune de ces familles portait sa propre variation génétique spécifique, et toutes suivaient un mode de transmission récessif : la maladie apparaissait seulement lorsque des individus héritaient de deux copies défectueuses, une de chaque parent. Dans une famille présentant des troubles de l’équilibre d’apparition adulte et des difficultés d’élocution, le responsable était une variation rare du gène MFSD8, qui participe au transport de matériaux vers des compartiments de recyclage cellulaires appelés lysosomes. Dans une autre famille, une mutation délétère dans AFG3L2, une protéine qui maintient la santé des mitochondries — les centrales énergétiques de la cellule — était associée à une ataxie spastique d’apparition infantile avec dystonie (contractions musculaires anormales). Une troisième famille portait une erreur de décalage du cadre de lecture (frameshift) dans SETX, un gène qui protège l’ADN lors de la réparation et est déjà connu pour provoquer une ataxie avec apraxie oculomotrice, un trouble affectant aussi les mouvements des yeux.
Un examen approfondi des atteintes nerveuses héréditaires
Deux autres familles présentaient une forme de maladie de Charcot‑Marie‑Tooth (CMT), un ensemble d’affections héréditaires qui endommagent les longs nerfs des pieds et des mains. Dans les deux cas, les chercheurs ont trouvé des variants délétères dans le gène GDAP1, crucial pour le fonctionnement normal des mitochondries dans les cellules nerveuses. Une altération de GDAP1 tronquait la protéine et était associée à une maladie très sévère d’apparition précoce ; une autre remplaçait un seul acide aminé et entraînait un tableau cliniquement plus modéré. Fait remarquable, le patient le plus gravement atteint dans une des familles CMT était également homozygote pour un variant pathogène connu dans un second gène, MMACHC, impliqué dans le métabolisme de la vitamine B12 et susceptible, dans certains cas, de répondre à des traitements à base de vitamines. Cette double atteinte pourrait expliquer pourquoi ses symptômes étaient plus sévères que ceux de ses apparentés ne portant pas le variant MMACHC.
Quand la recherche dans l’ADN ne suffit pas
Toutes les familles n’ont pas donné de réponse génétique claire. Dans deux des sept familles, l’équipe n’a pas identifié de variation unique dans l’exome correspondant de manière convaincante au schéma de transmission de la maladie. Dans un cas, ils ont repéré une variante du gène EHHADH qui suivait le mode de transmission mais est prédite bénigne et est connue pour causer une autre affection rénale lorsqu’elle est altérée. Dans un autre cas, deux cousins présentant des troubles du mouvement similaires se sont révélés avoir des causes sous‑jacentes différentes : un garçon portait un variant pathogène connu dans ALS2, qui peut mener à des formes juvéniles de maladie du motoneurone, tandis que ses cousins affectés n’en étaient pas porteurs. Ces cas non résolus suggèrent que des mutations importantes peuvent se trouver dans des régions du génome non couvertes par le séquençage de l’exome standard, ou que plusieurs facteurs génétiques subtils interagissent.
Ce que cela signifie pour les patients et les soins futurs
Globalement, les résultats montrent que les outils génétiques puissants peuvent dévoiler les gènes précis responsables de troubles complexes de l’équilibre et des nerfs, même dans des contextes de ressources limitées. Pour les cinq familles avec des conclusions claires, le travail transforme des étiquettes vagues comme « ataxie » ou « neuropathie » en diagnostics précis liés à des gènes spécifiques, ce qui peut orienter le conseil génétique, informer le pronostic et, dans certains cas, mettre en lumière des options thérapeutiques, comme des traitements liés à la vitamine B12 pour les maladies associées à MMACHC. L’étude enrichit aussi la compréhension des chercheurs sur la façon dont des gènes tels que MFSD8, AFG3L2, SETX, GDAP1, MMACHC et ALS2 influencent la santé des neurones du cerveau, de la moelle épinière et des nerfs périphériques. À l’avenir, des séquençages génomiques plus complets et des études fonctionnelles seront nécessaires pour résoudre les mystères restants et traduire ces connaissances génétiques en diagnostics plus précoces et en meilleurs soins pour les enfants et les adultes concernés. 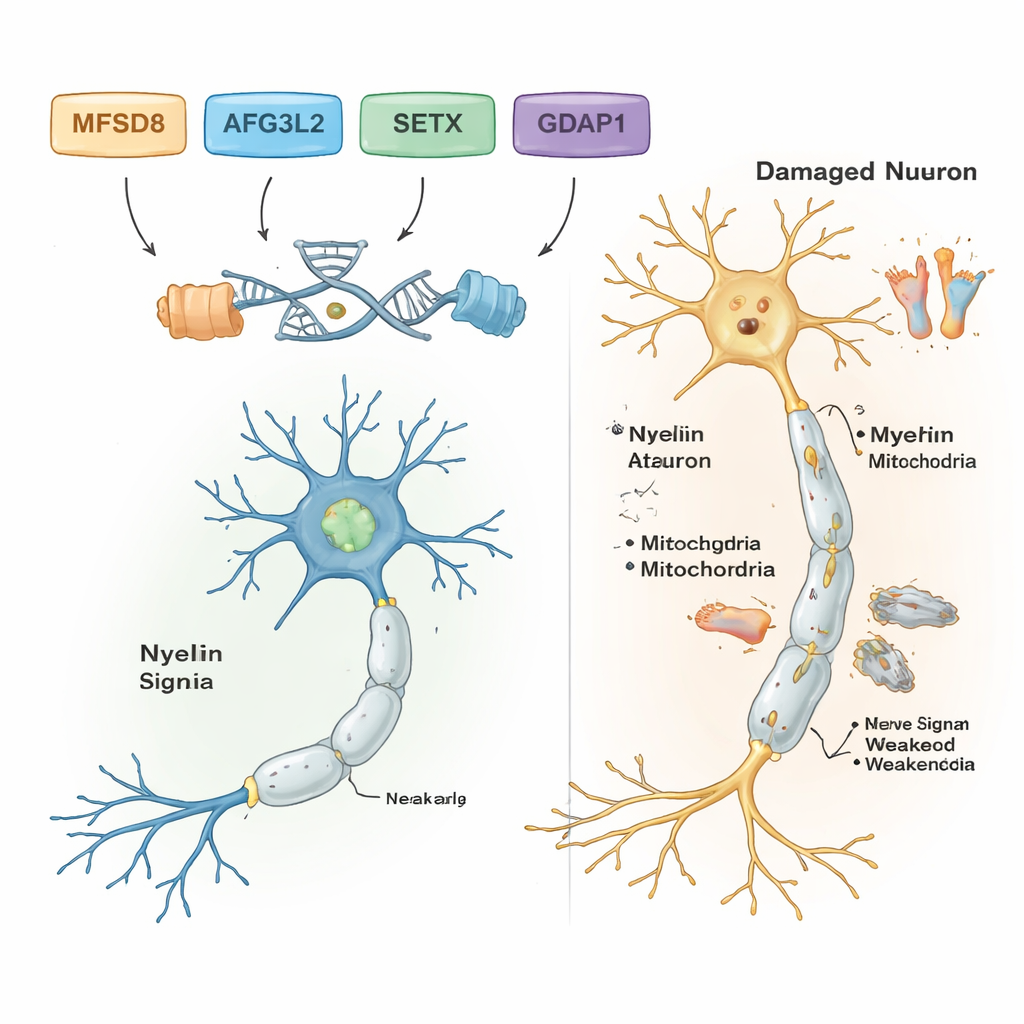
Citation: Aslam, F., Wajid, M., Butt, A.I. et al. Molecular characterization of recessively inherited ataxic and neuropathic disorders in consanguineous Pakistani families. Sci Rep 16, 6529 (2026). https://doi.org/10.1038/s41598-026-37808-0
Mots-clés: ataxie, neuropathie périphérique, séquençage de l’exome, maladie de Charcot‑Marie‑Tooth, diagnostic génétique