Clear Sky Science · fr
Optimisation de la sélection de caractéristiques dans les données de microarray cancéreux à l’aide d’un cadre évolutionniste guidé par un tas pour les espaces de haute dimension
Pourquoi choisir les bons gènes est important
Les tests contre le cancer basés sur les technologies génétiques modernes peuvent mesurer des dizaines de milliers de gènes en une seule fois, mais les médecins disposent souvent de données provenant de seulement quelques dizaines de patients. Cachés dans cette énorme « jungle génétique » se trouvent un nombre beaucoup plus restreint de signaux qui distinguent réellement un type de cancer d’un autre, ou une tumeur d’un tissu sain. Cet article présente une nouvelle méthode de recherche intelligente pour sélectionner automatiquement ces gènes clés, visant à rendre le diagnostic assisté par ordinateur plus précis, plus rapide et plus facile à interpréter.
Trop de signaux, trop peu de données
Les expériences de microarray et des technologies similaires permettent aux chercheurs de mesurer les niveaux d’expression de milliers de gènes pour chaque échantillon de patient. Pourtant, le nombre d’échantillons est généralement très réduit, parfois inférieur à une centaine. Beaucoup de ces mesures génétiques sont bruitées, redondantes ou sans rapport avec la maladie étudiée. Les conserver toutes peut submerger les algorithmes d’apprentissage, ralentir les calculs et produire des modèles trompeurs qui se fixent sur des particularités aléatoires plutôt que sur la biologie réelle. Le processus qui consiste à réduire cela à un sous-ensemble utile s’appelle « sélection de caractéristiques », et il est crucial si l’on veut des prédictions fiables à partir de données médicales de haute dimension.
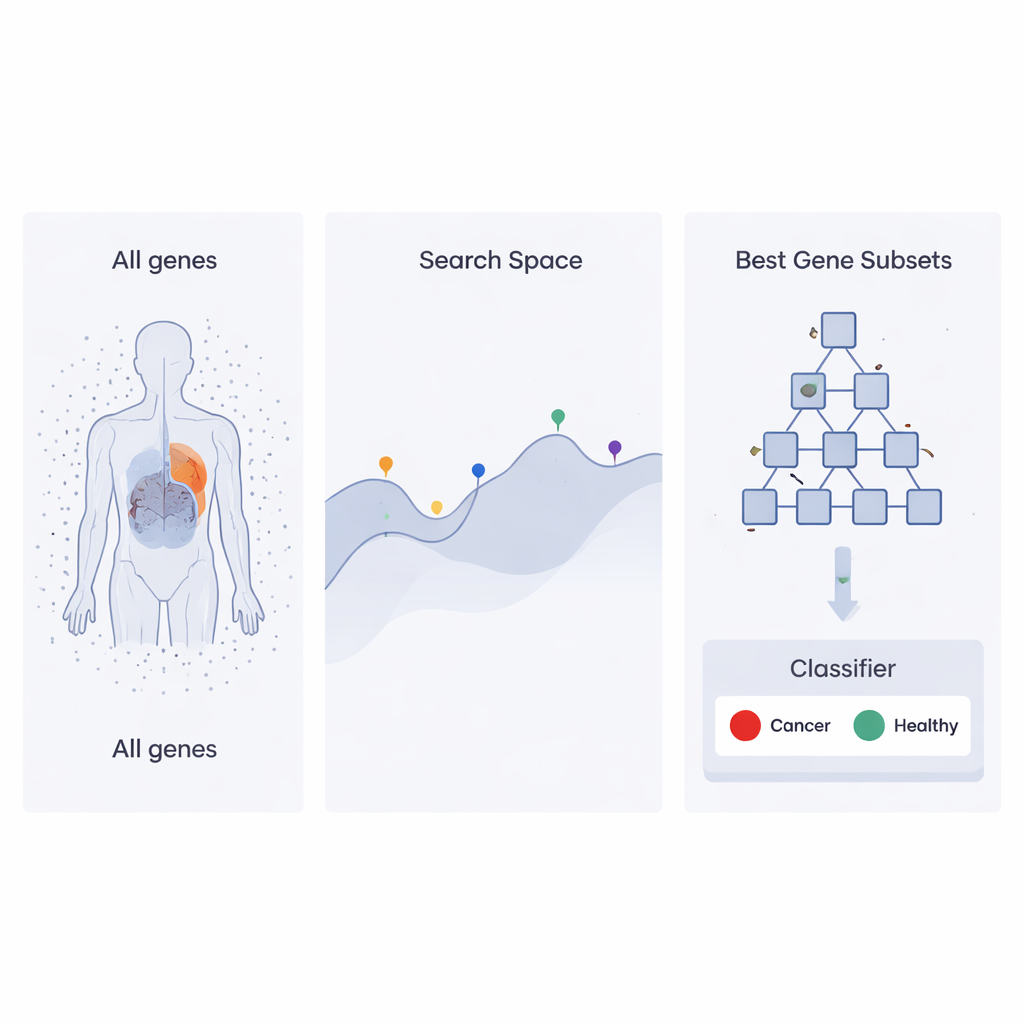
Une stratégie de recherche inspirée des échelles hiérarchiques en entreprise
Les auteurs s’appuient sur une approche d’optimisation récente nommée Heap‑Based Optimizer (HBO), qui emprunte des idées à la façon dont les employés sont organisés dans une entreprise. Imaginez chaque ensemble possible de gènes comme un « employé » dont la performance est jugée par sa capacité à aider un classifieur à distinguer des échantillons cancéreux des échantillons sains. Ces « employés » sont disposés dans une hiérarchie, comme une échelle d’entreprise, en utilisant une structure informatique connue sous le nom de tas (heap). Les ensembles de gènes les plus performants se trouvent près du sommet, tandis que les plus faibles sont plus bas. Au fil de nombreuses itérations, les éléments de rang inférieur ajustent leurs choix en copiant et en modifiant légèrement ce que font leurs supérieurs et collègues, poussant progressivement l’ensemble de l’organisation vers de meilleures solutions.
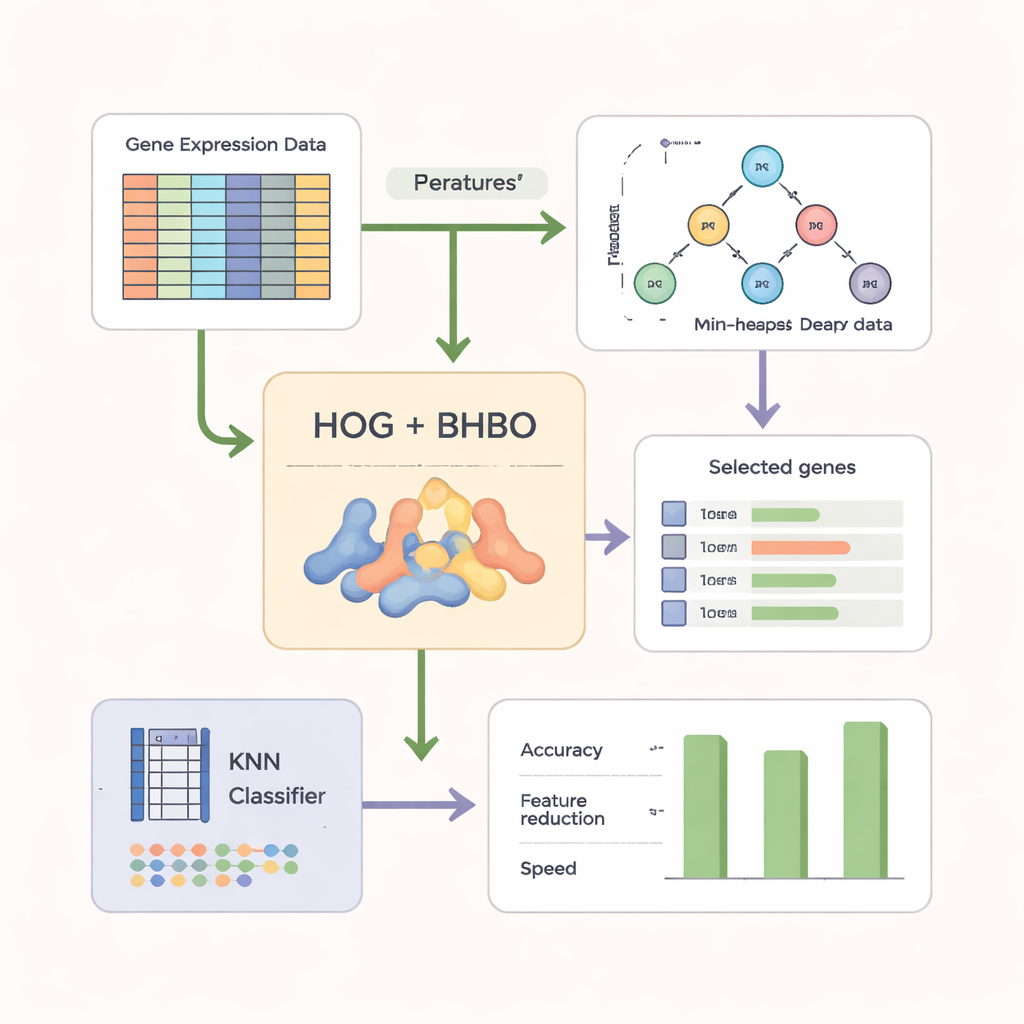
Transformer les données brutes en motifs plus nets
Pour rendre la recherche plus efficace, les auteurs ne se contentent pas des lectures brutes des gènes. Ils reformatent d’abord les données de microarray sous une forme proche d’une image et appliquent une technique appelée Histogram of Oriented Gradients (HOG), largement utilisée en vision par ordinateur. HOG capture la façon dont les niveaux d’expression varient à travers les gènes, mettant en évidence des motifs locaux plutôt que des mesures isolées. Ces caractéristiques basées sur des motifs sont ensuite combinées avec l’information génétique d’origine. Un classifieur simple appelé k‑Nearest Neighbors (KNN) sert de « juge », évaluant chaque sous-ensemble de gènes candidat en fonction de la précision avec laquelle il étiquette de nouveaux échantillons tout en récompensant les ensembles plus petits et plus compacts.
Tests sur plusieurs jeux de données cancéreuses
Les chercheurs ont évalué leur version binaire du Heap‑Based Optimizer (BHBO) sur neuf jeux de données publics de microarray cancéreux, incluant tumeurs cérébrales, leucémies, cancer de la prostate et collections de tumeurs mixtes avec de nombreux sous‑types. Chaque jeu de données comprenait des milliers à plus de quinze mille gènes mesurés mais relativement peu d’échantillons patients. Pour chaque jeu, BHBO a été exécuté à plusieurs reprises et comparé à sept méthodes de recherche bien connues, telles que les algorithmes génétiques et l’optimisation par essaim de particules. L’équipe a mesuré non seulement la précision, mais aussi le nombre de gènes conservés, la rapidité de convergence de la recherche et la stabilité des résultats lorsque les données étaient perturbées par du bruit simulé, des effets de lot et des erreurs d’étiquetage.
Les performances atteintes par la nouvelle méthode
Sur les neuf jeux de données, l’approche guidée par le tas a atteint une précision moyenne de classification d’environ 95 % tout en réduisant le nombre de gènes de plus de 85 %. Elle a nettement surpassé les méthodes concurrentes sur plusieurs jeux de données et montré une convergence plus rapide, c’est‑à‑dire qu’elle a trouvé de bons ensembles de gènes en moins d’itérations. Même lorsque les auteurs ont volontairement corrompu les données — en ajoutant du bruit ou en inversant certaines étiquettes d’échantillons — les performances de la méthode n’ont que légèrement diminué et sont restées supérieures aux alternatives. Des tests statistiques ont confirmé que ces gains étaient peu susceptibles d’être dus au hasard.
Ce que cela signifie pour les diagnostics du cancer à venir
Concrètement, ce travail montre qu’une stratégie de recherche bien conçue peut tamiser d’énormes jeux de données génétiques et découvrir de petits panels de gènes riches en information qui permettent encore de bien classifier les cancers. Pour les cliniciens et les chercheurs, ces ensembles compacts de gènes sont plus faciles à valider biologiquement, moins coûteux à mesurer lors de tests de suivi et mieux adaptés à l’intégration dans des outils d’aide à la décision. Bien que la méthode ne découvre pas directement de nouveaux médicaments ou voies biologiques, elle met en lumière des marqueurs génétiques prometteurs, aidant d’autres études à se concentrer sur les signaux les plus informatifs cachés dans les données cancéreuses de haute dimension.
Citation: Alweshah, M., Jebril, H., Kassaymeh, S. et al. Optimizing feature selection in cancer microarray data using a heap-driven evolutionary framework for high-dimensional spaces. Sci Rep 16, 6726 (2026). https://doi.org/10.1038/s41598-026-37803-5
Mots-clés: microarray cancéreux, sélection de caractéristiques, optimisation métaheuristique, biomarqueurs géniques, fouille de données médicales