Clear Sky Science · fr
Stratégies computationnelles pour extraire des informations à partir d’inhibiteurs connus en vue d’une optimisation des leads : étude de cas sur des analogues du célécoxib
Pourquoi de petits changements dans les antalgiques comptent
Les analgésiques modernes n’atténuent pas seulement la douleur ; ils modifient la chimie du corps de manière très précise. Le célécoxib, un anti‑inflammatoire répandu, cible une enzyme impliquée dans la douleur et l’inflammation tout en épargnant en grande partie une enzyme apparentée qui protège l’estomac. Pourtant, des dizaines de cousins chimiques proches du célécoxib se comportent très différemment dans l’organisme. Cette étude utilise la modélisation informatique pour poser une question apparemment simple mais aux grandes implications pour des médicaments plus sûrs : dans quelle mesure un petit changement dans une molécule a‑t‑il de l’importance ?
L’enzyme qui déclenche la douleur
Lorsque le tissu est blessé ou enflammé, l’organisme libère une molécule grasse appelée acide arachidonique. Une enzyme nommée COX‑2 convertit cette molécule en prostaglandines, qui déclenchent douleur, fièvre et gonflement. Une enzyme apparentée, COX‑1, contribue à protéger la muqueuse gastrique et les plaquettes. Les anciens analgésiques comme l’ibuprofène atteignent les deux enzymes, soulagent la douleur mais irritent souvent l’intestin. Le célécoxib a été conçu pour se glisser dans un creux légèrement plus grand présent principalement dans la COX‑2, bloquant les signaux de douleur tout en laissant intactes une grande partie des fonctions protectrices de la COX‑1. Comprendre la forme détaillée de ce creux, et comment les molécules médicamenteuses s’y positionnent, est central pour concevoir de nouveaux médicaments à la fois puissants et sûrs.
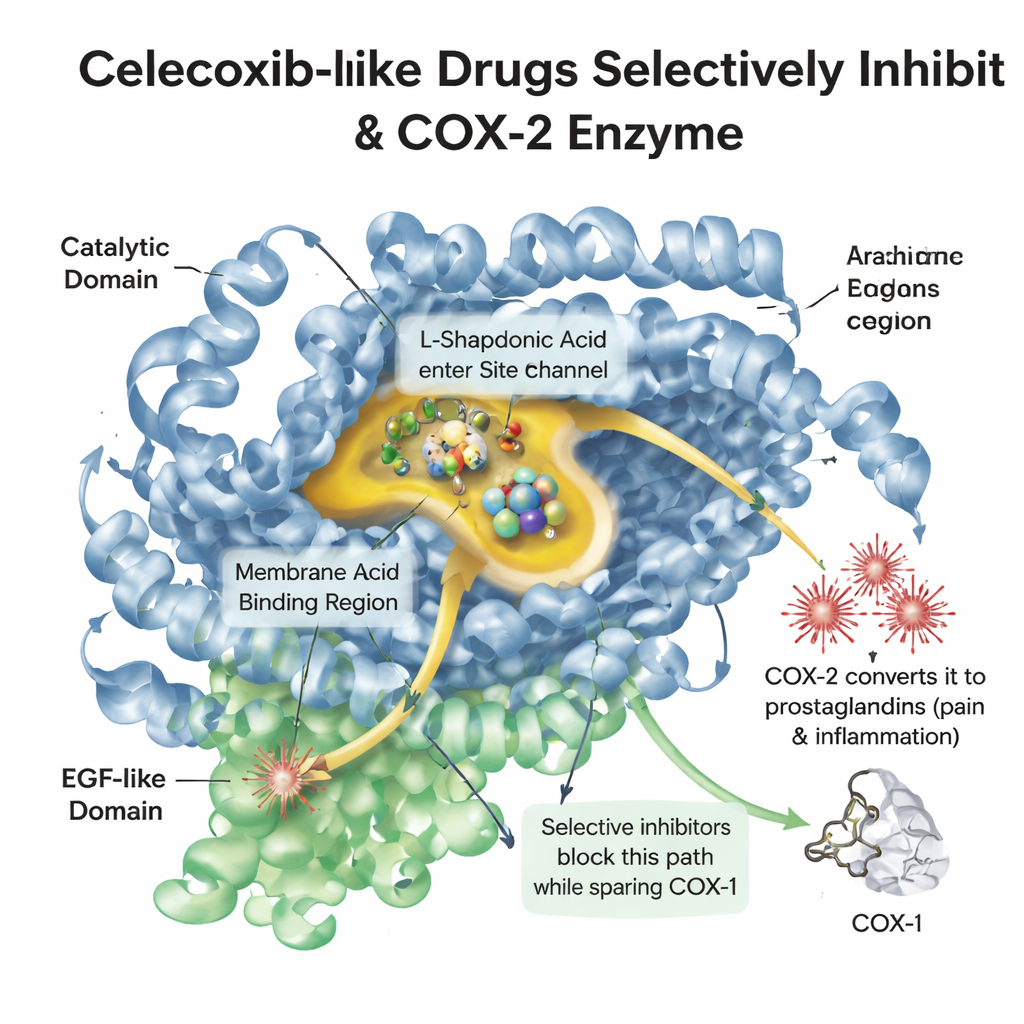
Une bibliothèque numérique de molécules ressemblantes
Les chercheurs ont rassemblé un jeu de 375 molécules qui partagent toutes le squelette à trois cycles du célécoxib mais diffèrent par de petites modifications, comme l’échange d’un seul atome ou d’un groupe latéral. Ils ont extrait ces structures et leurs forces d’inhibition de la COX‑2 mesurées à partir d’une base de données publique sur les médicaments. À l’aide de logiciels de chimie, ils ont généré des modèles 3D de chaque molécule, calculé près de 2 000 descripteurs numériques de leurs formes et propriétés, puis les ont dockées dans une structure haute résolution de l’enzyme COX‑2. Lors du docking, un ordinateur place la molécule de différentes façons dans le creux de l’enzyme et évalue à quel point chaque position s’adapte bien.
Ce qui contrôle vraiment la puissance et la sélectivité
L’équipe s’est concentrée sur trois régions clés du célécoxib. « Site‑1 » est un cycle qui se loge dans une zone hydrophobe du creux ; « Site‑2 » est un cycle portant une queue riche en fluor ; et « Site‑3 » est un cycle portant un groupe sulfonamide qui forme de fortes liaisons hydrogène. Leur analyse a montré que le Site‑1 préfère des groupes petits et non polaires qui préservent les contacts hydrophobes ; rendre cette région plus hydrophile, par exemple en ajoutant un –OH ou un groupe acide, affaiblissait généralement le médicament. Au Site‑2, de petits groupes électronégatifs tels que le fluor affinaient souvent la puissance en améliorant les interactions dans un creux étroit, tandis que des queues plus volumineuses ou plus polaires nuisaient à l’activité. Au Site‑3, l’azote du sulfonamide, capable de donner une liaison hydrogène, était crucial ; le remplacer par une version non liaisonnante réduisait sensiblement l’affinité.
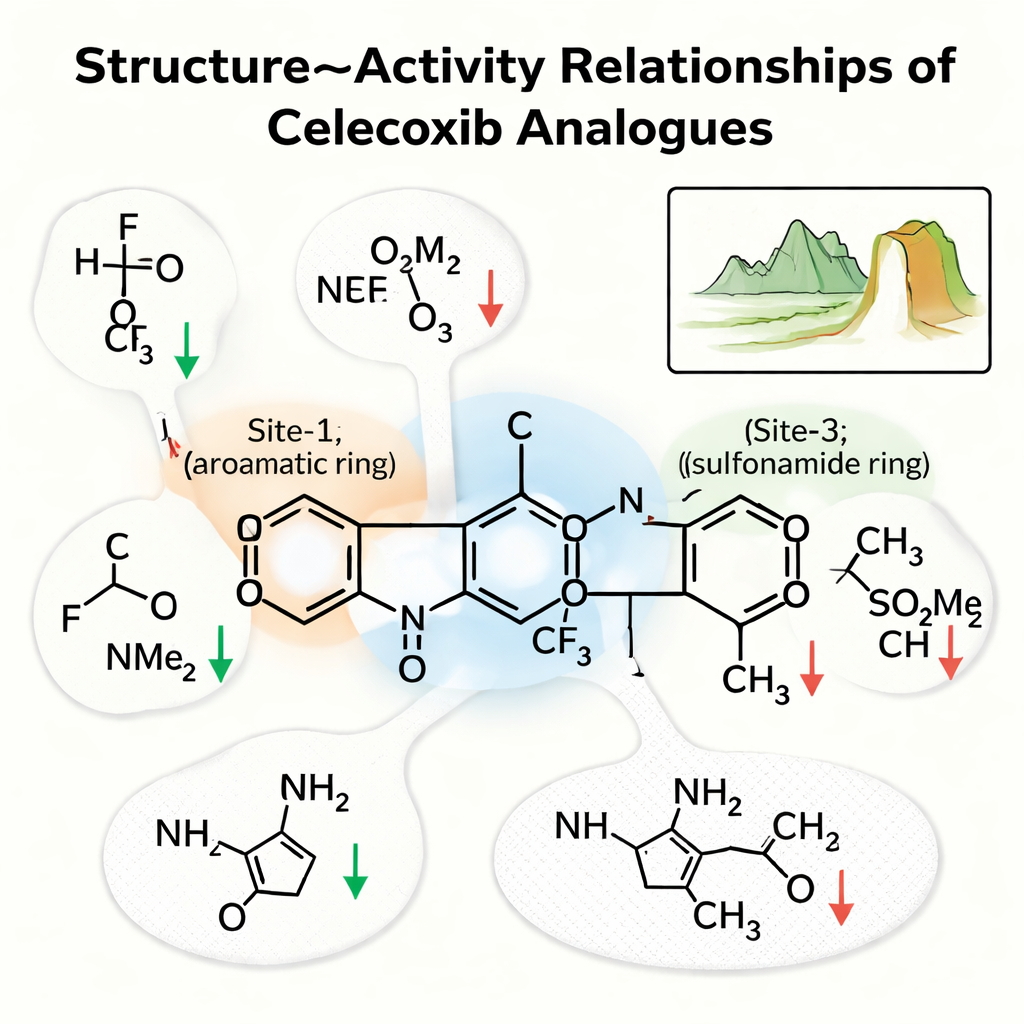
Falaises dans le paysage chimique
Pour aller au‑delà des tendances simples, les auteurs ont construit un « paysage structure‑activité », qui interroge combien la puissance du médicament change lorsque la structure varie très peu. Dans cette optique, la plupart des composés de type célécoxib se situent sur des collines douces : les ajuster — par exemple en déplaçant un halogène ou en ajoutant un petit groupe flexible — augmente ou diminue la puissance de manière prévisible. Mais quelques paires forment de nettes « falaises d’activité », où un minuscule changement, comme remplacer un groupe méthyle par un groupe trifluorométhyle ou ajouter un seul atome de fluor, provoque un gain ou une perte d’activité spectaculaire. L’étude a également réalisé des simulations de dynamique moléculaire complètes — des films virtuels des complexes médicament‑enzyme en mouvement — qui ont confirmé que les meilleurs analogues se maintiennent de façon stable dans le creux pendant des centaines de nanosecondes.
Orienter la prochaine génération d’antalgiques plus sûrs
Pour un non‑spécialiste, le message essentiel est que, en conception de médicaments, les petits détails comptent énormément. Deux composés qui semblent presque identiques sur le papier peuvent différer d’un facteur mille dans leur capacité à bloquer la COX‑2, simplement parce qu’un atome supplémentaire améliore l’emboîtement dans un creux microscopique ou perturbe un contact clé. En cartographiant systématiquement quelles modifications aident ou nuisent à chacun des trois sites clés du célécoxib, et en mettant en évidence les « falaises » dangereuses où de minuscules ajustements ont des effets disproportionnés, ce travail computationnel offre une feuille de route pour les chimistes. Il indique des pistes pour de nouveaux anti‑inflammatoires qui conservent l’efficacité analgésique du célécoxib tout en améliorant encore la sécurité et la sélectivité.
Citation: Grewal, S., Ghosh, B., Narayan, U. et al. Computational strategies for unraveling insights from known inhibitors for further lead optimization: A case study on Celecoxib analogues. Sci Rep 16, 6720 (2026). https://doi.org/10.1038/s41598-026-37798-z
Mots-clés: inhibiteurs de la COX-2, analogues du célécoxib, médicaments anti-inflammatoires, conception de médicaments par calcul, relations structure-activité