Clear Sky Science · fr
Une méthode de détection des variants structuraux utilisant la matrice de contacts Hi-C et des réseaux neuronaux
Pourquoi la courbure de l’ADN en 3D compte
Notre ADN est généralement représenté comme une simple chaîne de lettres, mais à l’intérieur de chaque cellule il se replie en une forme tridimensionnelle complexe. Lorsque de gros segments de cette chaîne sont supprimés, inversés ou déplacés — des modifications appelées variations structurales — ils peuvent perturber des gènes et favoriser le développement du cancer. Cette étude présente VarHiCNet, un nouveau système d’intelligence artificielle qui lit les cartes de repliement 3D de l’ADN et repère ces changements à grande échelle plus précisément que les outils existants, offrant une nouvelle façon d’étudier les génomes cancéreux et d’autres maladies.
Voir les changements du génome via des cartes de contacts 3D
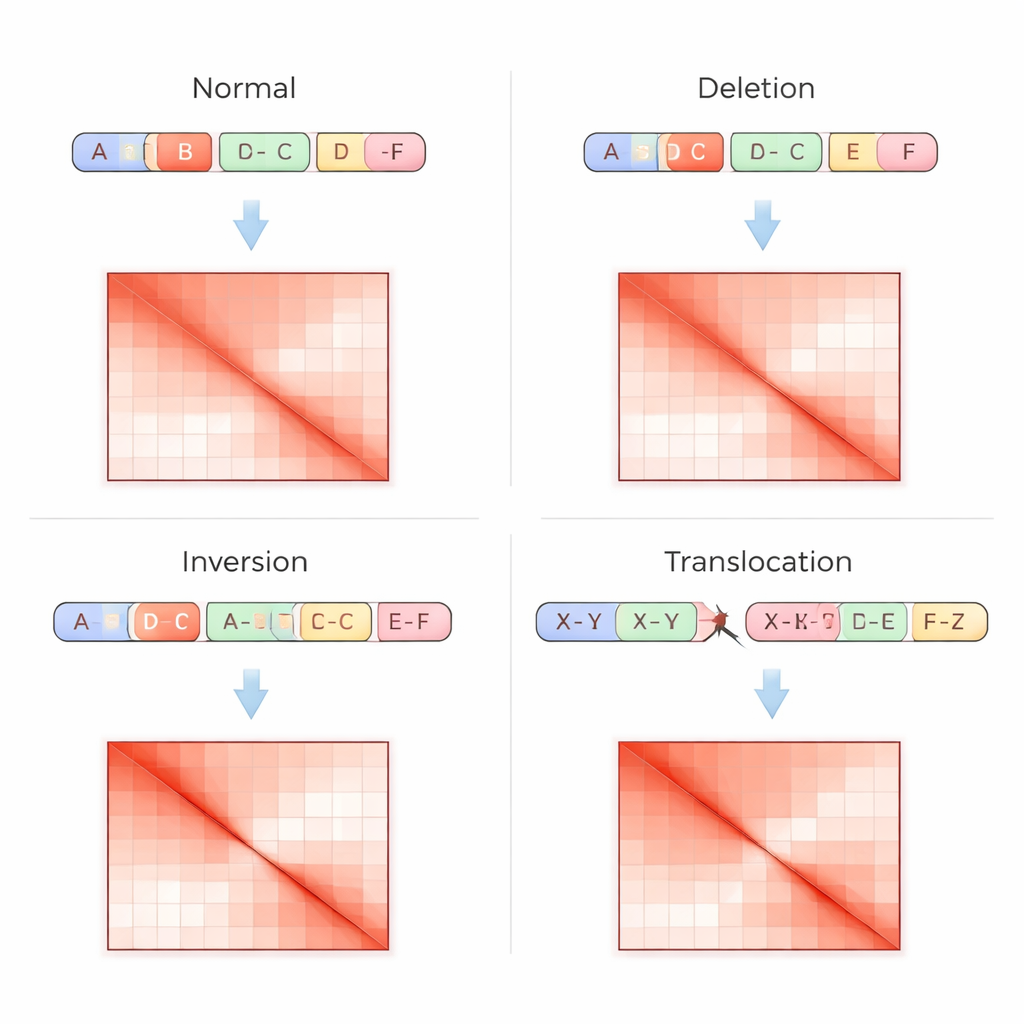
Les tests génomiques traditionnels lisent l’ADN comme une séquence linéaire, ce qui rend difficile la détection de réarrangements complexes, notamment dans les régions répétitives ou lorsque des segments sont déplacés sans changer leur nombre de copies. La technique Hi-C aborde le problème différemment : elle mesure la fréquence à laquelle des portions éloignées de l’ADN se touchent physiquement dans le noyau, puis enregistre ces contacts sous forme d’une grille, ou matrice de contacts, où des taches plus lumineuses indiquent des interactions plus fortes. Les variations structurales laissent des empreintes caractéristiques dans ces matrices — comme des bandes manquantes lorsqu’une région est supprimée, des motifs miroir lorsqu’un segment est inversé, ou des points chauds hors diagonale lorsqu’il y a fusion entre deux chromosomes. VarHiCNet est conçu pour reconnaître automatiquement ces motifs visuels.
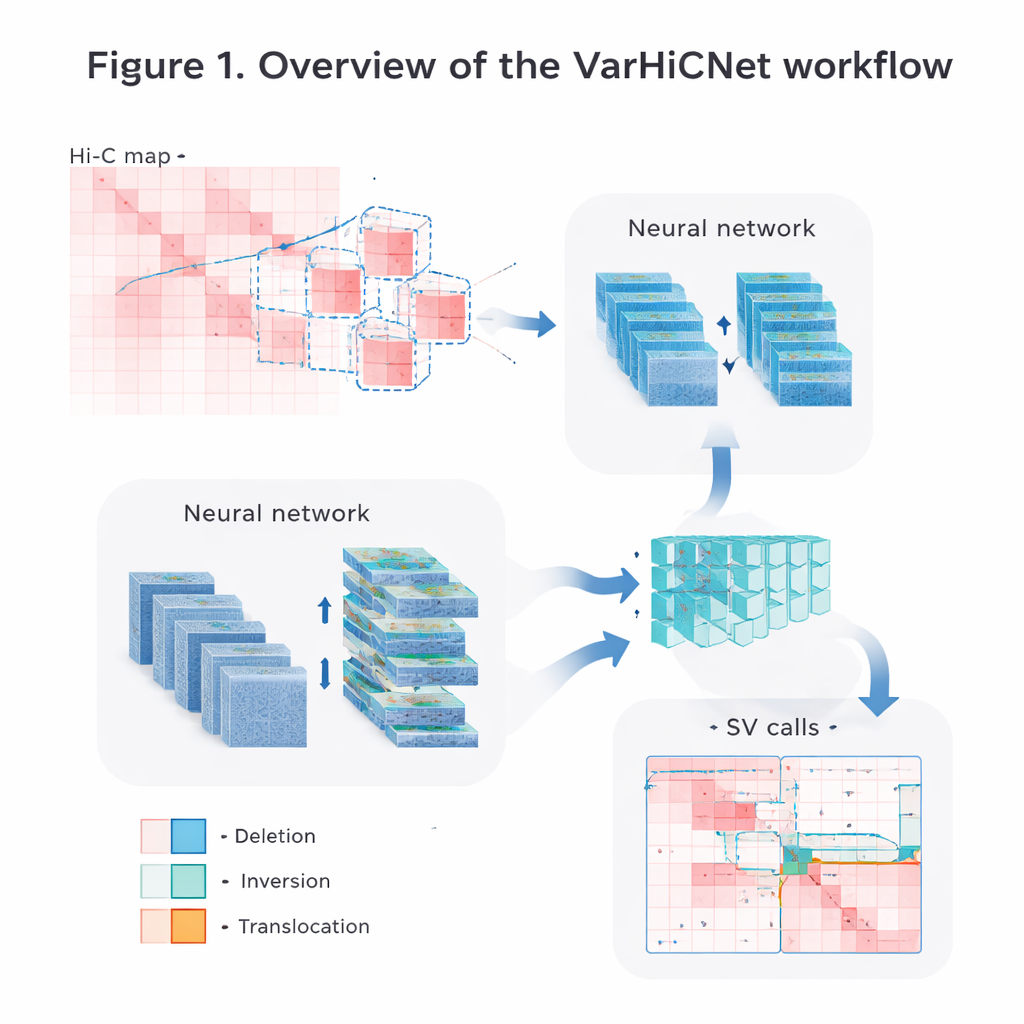
Transformer des cartes du génome en images pour l’IA
Les auteurs convertissent les données brutes de contacts Hi-C en images que les systèmes de vision par ordinateur peuvent traiter facilement. D’abord, ils normalisent soigneusement les matrices pour corriger la décroissance naturelle de la fréquence de contact à mesure que les segments d’ADN s’éloignent, tout en préservant les signaux d’interaction à courte et longue portée. Ensuite, ils parcourent chaque chromosome avec des fenêtres carrées chevauchantes et découpent de nombreuses sous-matrices plus petites. Chaque sous-matrice est redimensionnée en une image couleur standardisée de 800 par 800 pixels, où différentes intensités de contact sont mappées en tons rouges sur trois canaux de couleur. Cette représentation de type image permet au modèle de réutiliser des techniques puissantes initialement développées pour la reconnaissance d’objets dans des photographies.
Emprunter des astuces à la détection d’objets
VarHiCNet traite chaque variant structurel potentiel comme s’il s’agissait d’un « objet » dans une image. Il s’appuie sur un cadre moderne de détection d’objets appelé RT-DETR, qui combine des réseaux de neurones convolutionnels et des Transformers pour mettre en évidence les régions importantes. Une colonne vertébrale ResNet extrait d’abord des caractéristiques multi-échelle : les couches peu profondes conservent le détail fin nécessaire pour localiser précisément les points de rupture, tandis que les couches profondes captent des motifs plus larges signalant des évènements d’envergure. Un module de fusion de caractéristiques mélange ensuite l’information de plusieurs couches afin que soient préservés à la fois les indices locaux et globaux. Un autre bloc personnalisé, inspiré par le regroupement en pyramide spatiale, ajuste la quantité de région environnante que le modèle « voit » à la fois, le rendant sensible à des variants couvrant aussi bien une étendue relativement petite qu’un très large segment d’ADN.
Des régions candidates aux types de variants précis
Une fois que VarHiCNet a proposé des régions candidates dans l’image Hi-C, il doit les affiner en points de rupture exacts et en types de variants spécifiques, tels que suppressions, inversions, duplications ou translocations. Pour ce faire, le système zoome sur le voisinage de chaque point de rupture prédit et en réduit la complexité avec une technique mathématique appelée analyse en composantes principales, qui met en évidence où le motif de contacts change le plus fortement. Ces représentations compactes sont ensuite introduites dans un classificateur basé sur un Transformer qui apprend les différences subtiles des motifs locaux pour chaque catégorie de variant. Le résultat est un appel détaillé pour chaque évènement : où il se produit dans le génome et quel type de changement structural il représente.
Performances sur diverses lignées cellulaires cancéreuses
Les chercheurs ont testé VarHiCNet sur des données Hi-C de six lignées cellulaires cancéreuses humaines différentes, couvrant des tumeurs du sang, du sein, du cerveau, du rein, du poumon et de la prostate. En utilisant un catalogue de variants structuraux à haute confiance comme référence, ils ont comparé leur méthode à plusieurs outils de pointe analysant aussi les données Hi-C. Tant pour les évènements intra-chromosomiques qu’inter-chromosomiques, VarHiCNet a généralement obtenu des scores F1 supérieurs ou comparables, ce qui signifie qu’il équilibre mieux sensibilité et précision que d’autres approches. Il s’est montré particulièrement performant pour détecter les translocations équilibrées et les inversions — des réarrangements qui laissent souvent peu de trace dans le séquençage ADN standard mais qui produisent des signatures claires dans le repliement 3D. Les auteurs ont aussi démontré que leurs choix de conception, comme la résolution des images et les modules de fusion de caractéristiques, amélioraient systématiquement les performances dans des tests contrôlés.
Ce que cela signifie pour la compréhension des maladies
Concrètement, VarHiCNet offre aux scientifiques une façon plus intelligente de « voir » comment le génome se replie en 3D et de repérer de larges réarrangements liés à des maladies qui pourraient être manqués par le séquençage conventionnel seul. En transformant des cartes de contacts complexes en images et en appliquant des réseaux neuronaux de type vision moderne, la méthode peut détecter et catégoriser de nombreux types de variations structurales avec une grande fiabilité sur différents types de cancers. Bien qu’elle peine encore avec certains changements très petits ou fortement imbriqués et qu’elle dépende de données d’entraînement riches, VarHiCNet ouvre la voie à un avenir où l’architecture du génome en 3D devient une composante courante de la lecture, de l’interprétation et, à terme, du ciblage des altérations génétiques à l’origine du cancer et d’autres maladies.
Citation: Shen, J., Wang, H., Zhai, H. et al. A method for structural variant detection using Hi-C contact matrix and neural networks. Sci Rep 16, 7324 (2026). https://doi.org/10.1038/s41598-026-37678-6
Mots-clés: variation structurale, Hi-C, apprentissage profond, génomique du cancer, génome 3D