Clear Sky Science · fr
Les pools membranaires plasmatiques d’APOL1 résistent à une dégradation protéique rapide
Pourquoi le « tour de passe‑passe » d’une protéine rénale est important
Une grande part des formes graves de maladie rénale chez les personnes d’ascendance africaine récente a été attribuée à deux variantes d’un même gène, APOL1. Pourtant, les chercheurs peinent encore à expliquer comment ce gène endommage les cellules rénales sans nuire à la plupart des porteurs. Cette étude pose une question apparemment simple mais aux grandes implications : une fois synthétisée à l’intérieur des cellules, combien de temps la protéine APOL1 persiste‑t‑elle et où est‑elle la plus stable ? Les réponses révèlent une personnalité surprenante — APOL1 est rapidement détruite à l’intérieur des cellules mais reste obstinément stable lorsqu’elle est intégrée à la surface extérieure de la cellule, un indice pouvant orienter des thérapies futures.
Un gène à double tranchant
Le gène APOL1 contribue à la protection humaine contre certains parasites, un avantage évolutif qui explique probablement la fréquence élevée de ses variantes à risque, appelées G1 et G2, dans les populations africaines. Malheureusement, les personnes qui héritent de deux copies de ces variantes présentent un risque fortement accru de troubles rénaux regroupés sous le terme de maladies rénales médiées par APOL1. Des travaux antérieurs ont montré que lorsque les niveaux d’APOL1 augmentent — souvent en réponse à l’inflammation — la protéine peut devenir toxique, en particulier dans les cellules rénales filtrantes sensibles appelées podocytes. Mais la plupart des études se sont concentrées sur ce qui active APOL1. On savait beaucoup moins comment les cellules l’éteignent à nouveau, par exemple en dégradant la protéine.
Suivre une protéine fragile à l’intérieur des cellules
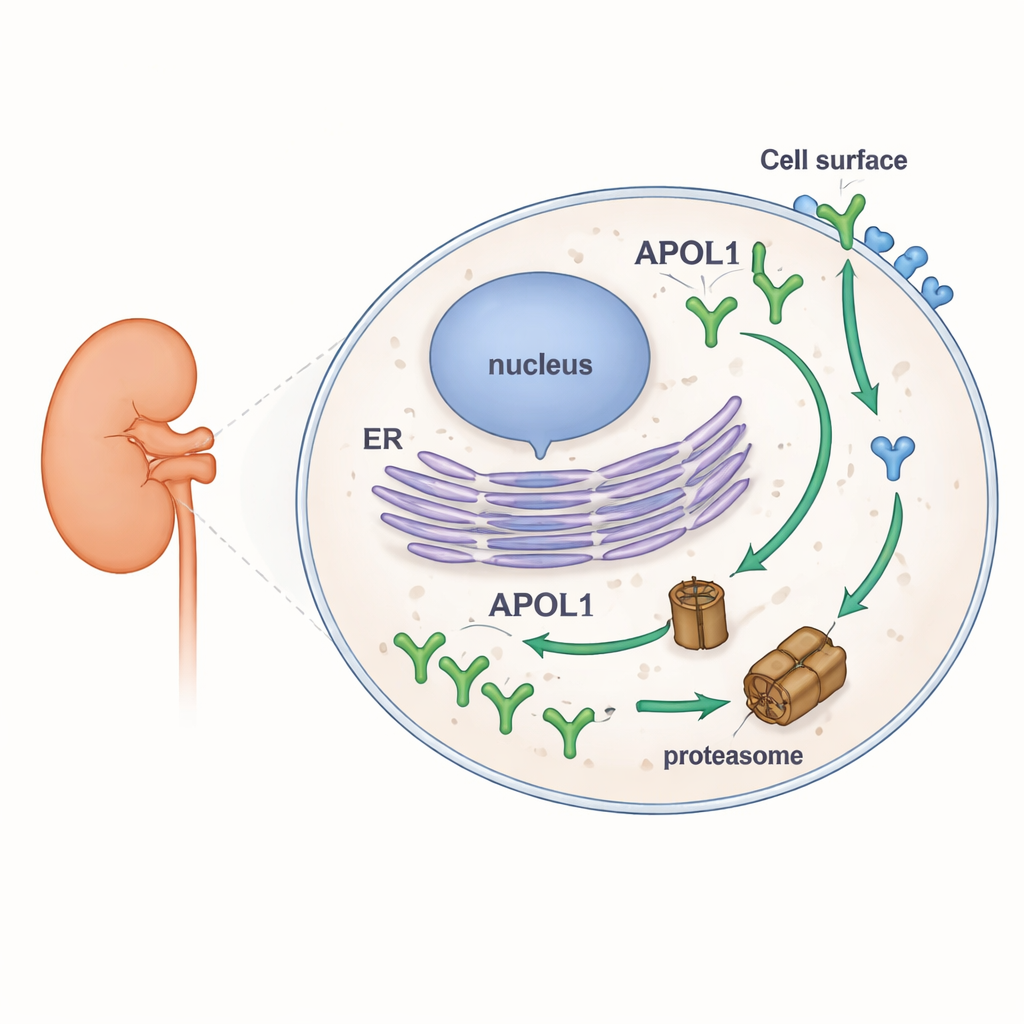
Pour explorer la stabilité d’APOL1, les auteurs ont modifié des lignées cellulaires humaines pour produire des versions fluorescentes d’APOL1 et de son plus proche homologues, APOL2. Cela leur a permis d’observer la quantité de chaque protéine accumulée ou perdue dans différentes conditions via le Western blot, la microscopie et la cytométrie en flux. Ils ont bloqué la principale machinerie intracellulaire de dégradation des protéines, le protéasome, puis, séparément, l’arrêt de la synthèse protéique. Lorsque le protéasome était inhibé, les niveaux d’APOL1 augmentaient rapidement, montrant qu’elle est normalement dégradée à un rythme élevé. Quand la synthèse protéique était arrêtée, les niveaux d’APOL1 chutaient rapidement. En nette opposition, APOL2 variait à peine sous l’un ou l’autre traitement, révélant une protéine beaucoup plus stable. Fait important, le fort renouvellement d’APOL1 était identique pour la version normale (G0) et pour les variantes à risque (G1 et G2), et se retrouvait dans plusieurs formes naturelles d’APOL1 qui diffèrent par leur insertion dans les membranes.
Indices de séquence et histoire de deux quartiers
En examinant la structure de la protéine, l’équipe a utilisé des outils informatiques pour rechercher chez APOL1 et APOL2 des segments lâches et non structurés, appelés régions intrinsèquement désordonnées. Ces régions fonctionnent souvent comme des signaux « mange‑moi » pour le protéasome. Ils ont identifié deux régions candidates marquées dans APOL1, largement absentes d’APOL2. Pour tester si l’extrémité avant unique d’APOL1 contribuait à sa fragilité, ils ont créé des hybrides : un APOL1 raccourci manquant ses 59 premiers acides aminés, et une chimère APOL2 portant ce segment d’APOL1. L’ajout du fragment N‑terminal d’APOL1 à APOL2 a rendu APOL2 plus susceptible à la dégradation, tandis que l’APOL1 tronquée restait instable, suggérant que plusieurs parties d’APOL1 favorisent la dégradation rapide. Ensemble, ces résultats lient les segments flexibles inhabituels d’APOL1 à son fort turnover, sans associer ce comportement spécifiquement aux variantes causant la maladie.
Une protéine tenace à la surface cellulaire
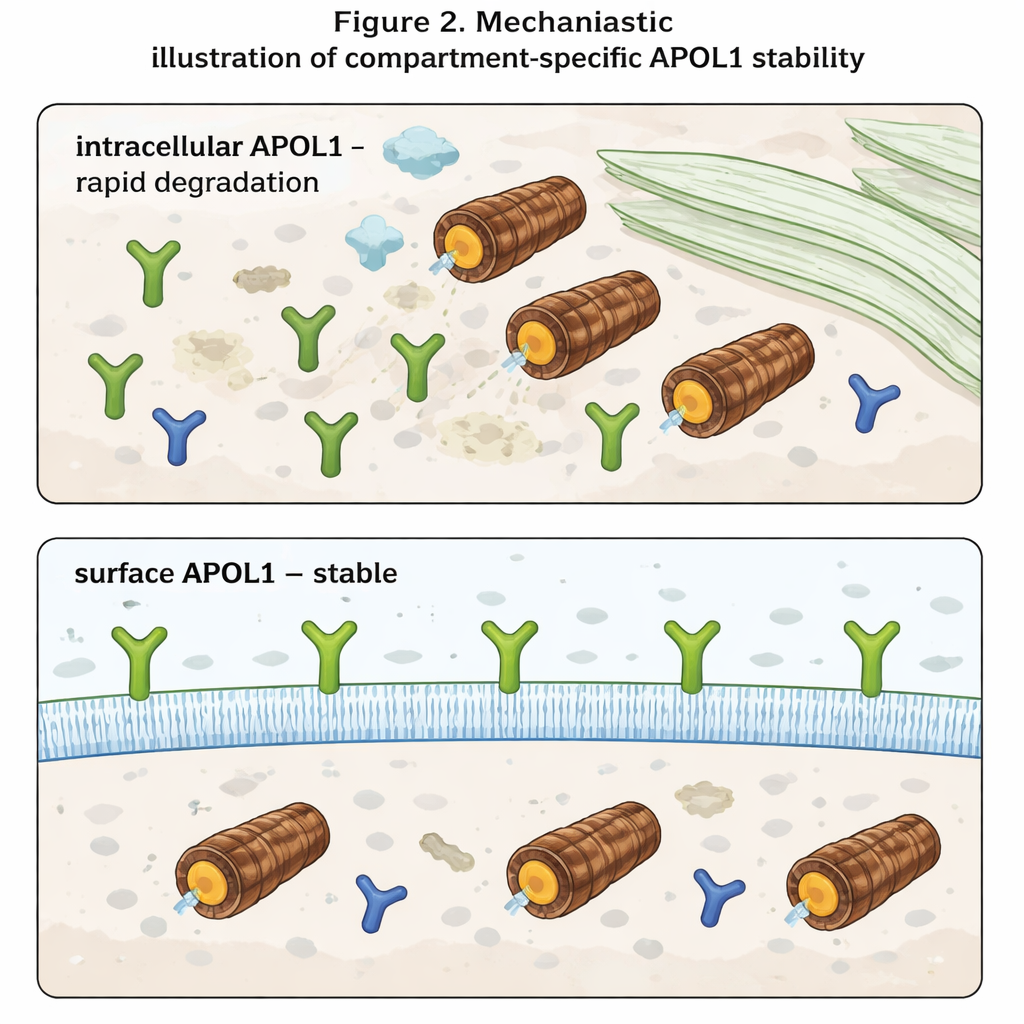
La découverte la plus marquante est apparue lorsque les auteurs ont distingué APOL1 intracellulaire et APOL1 à la surface cellulaire. À l’aide d’anticorps ne reconnaissant que la portion d’APOL1 exposée vers l’extérieur, ils ont mesuré séparément les niveaux de surface et les niveaux totaux. Dans la cellule, APOL1 se comportait comme prévu : elle s’accumulait quand le protéasome était bloqué et disparaissait rapidement quand la synthèse était arrêtée. L’APOL1 de surface, en revanche, bougeait très peu dans l’une ou l’autre condition. Une fois que les molécules d’APOL1 atteignaient la membrane plasmique, elles se sont avérées très résistantes à une dégradation rapide. De plus, bien que les variantes à risque produisent moins d’APOL1 total que la version normale, leurs niveaux de surface étaient similaires. Cela suggère que les variantes à risque et la version normale sont éliminées à des rythmes comparables à l’intérieur de la cellule, mais que les pools intégrés à la membrane — qui formeraient des canaux ioniques et contribueraient à la toxicité — sont préservés dans toutes les variantes.
Ce que cela signifie pour les traitements futurs
Pour les non‑spécialistes, la conclusion est que le comportement d’APOL1 dépend fortement de son emplacement. À l’intérieur de la cellule, c’est une protéine éphémère, rapidement reconnue et détruite. À la surface cellulaire, elle devient de longue durée et relativement protégée, même lorsque la machinerie de dégradation de la cellule est modifiée. Puisque la maladie semble émerger lorsque les canaux APOL1 à la surface perturbent l’équilibre d’ions comme le sodium et le potassium, les stratégies thérapeutiques pourraient devoir se concentrer moins sur les niveaux totaux d’APOL1 que sur la quantité qui atteint et persiste dans la membrane plasmique. Des approches réduisant le trafic d’APOL1 vers la surface ou déstabilisant sélectivement le pool de surface pourraient, en principe, atténuer les lésions rénales sans bloquer complètement les fonctions immunitaires bénéfiques du gène.
Citation: Höffken, V., Alvermann, L., Niggemeier, D. et al. APOL1 plasma membrane pools resist rapid protein degradation. Sci Rep 16, 6718 (2026). https://doi.org/10.1038/s41598-026-37647-z
Mots-clés: APOL1, maladie rénale, dégradation des protéines, membrane plasmique, protéasome