Clear Sky Science · fr
Caractérisation in vitro du domaine catalytique de l’histone désacétylase 5 humaine
Pourquoi de petits interrupteurs dans l’emballage de notre ADN comptent
À l’intérieur de chaque cellule, notre ADN est enroulé autour de protéines qui jouent le rôle de bobines, permettant d’empaqueter des mètres de matériel génétique dans un espace microscopique. L’allumage ou l’extinction d’un gène dépend souvent de petites étiquettes chimiques apposées sur ces protéines-bobines. Cette étude se concentre sur un « interrupteur » protéique particulier, appelé HDAC5, associé aux maladies cardiaques, aux troubles cérébraux, au cancer et à d’autres pathologies. Comprendre le fonctionnement d’HDAC5 au niveau moléculaire vise à ouvrir la voie à des médicaments plus précis et avec moins d’effets secondaires.
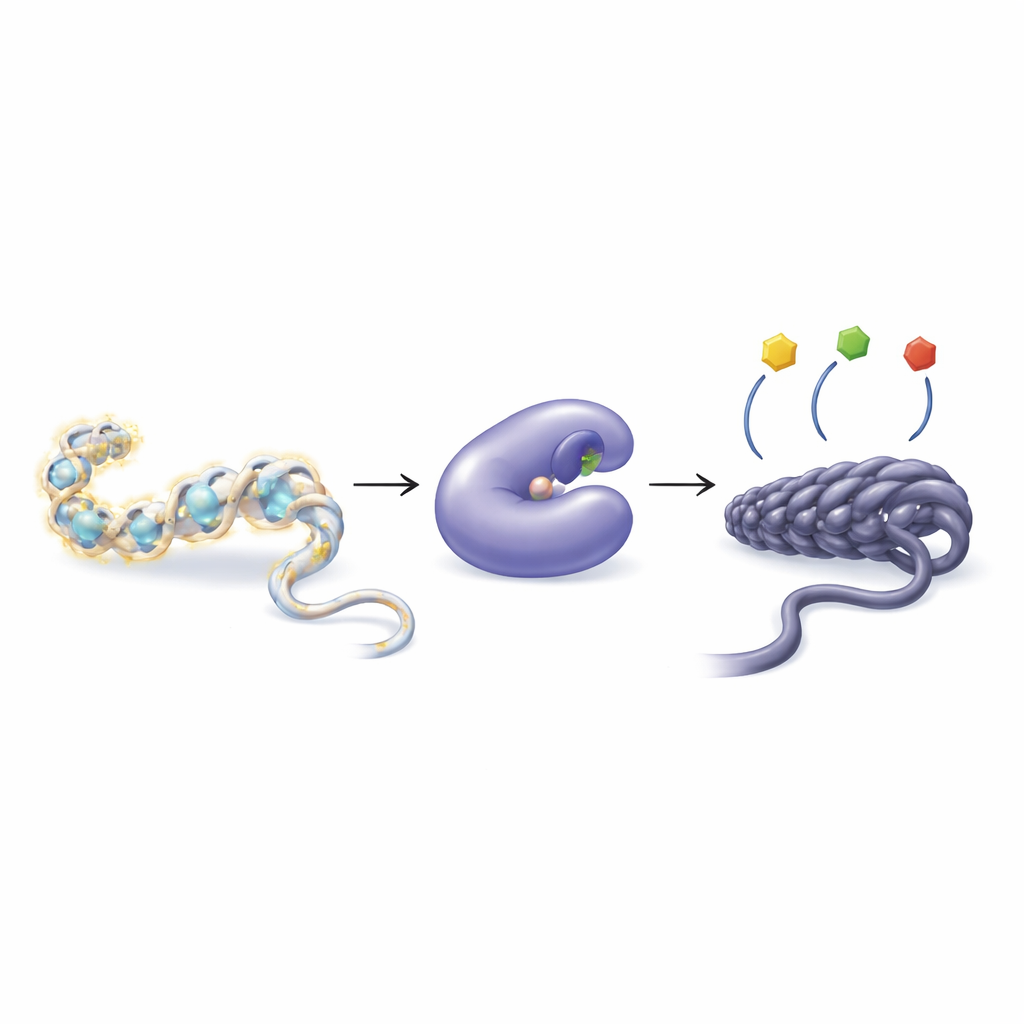
Comment les cellules règlent les gènes avec de petites étiquettes chimiques
Notre ADN ne flotte pas librement : il est enroulé autour de protéines appelées histones, formant une structure nommée chromatine. Les cellules peuvent ajouter ou retirer de petits groupes chimiques, comme des groupes acétyle, sur les queues des histones pour desserrer ou resserrer la chromatine. Un empaquetage lâche facilite généralement la lecture des gènes ; un empaquetage serré tend à les silencer. Deux classes d’enzymes gèrent cet équilibre : les histone acétyltransférases ajoutent des groupes acétyle, tandis que les histone désacétylases (HDAC) les retirent. Quand cet équilibre est perturbé, cela peut contribuer à de nombreuses maladies, dont le cancer, les problèmes cardiaques, la fonte musculaire et les troubles du système immunitaire.
Pourquoi HDAC5 est une cible médicamenteuse prometteuse mais complexe
Les HDAC forment une grande famille d’enzymes apparentées, divisée en plusieurs classes. De nombreux médicaments actuellement en clinique bloquent plusieurs types d’HDAC à la fois, ce qui peut interrompre des fonctions normales importantes et provoquer des effets secondaires marqués. Les HDAC de classe IIa, dont HDAC5, se distinguent parce qu’elles sont enrichies dans des tissus spécifiques comme le cerveau, le cœur et le muscle squelettique, et qu’elles s’associent à d’autres protéines pour réguler des réseaux de gènes clés. HDAC5 agit souvent comme un pont, amenant un partenaire enzymatique très actif (HDAC3) vers certains gènes afin de resserrer la chromatine et de réprimer ces gènes. En raison de ces rôles ciblés, HDAC5 apparaît comme une cible attractive pour des médicaments plus sélectifs, mais il manquait des données biochimiques détaillées et il n’existait pas de structure haute résolution de son cœur actif, ce qui compliquait la conception rationnelle de médicaments.
Reconstruire HDAC5 dans un tube à essai
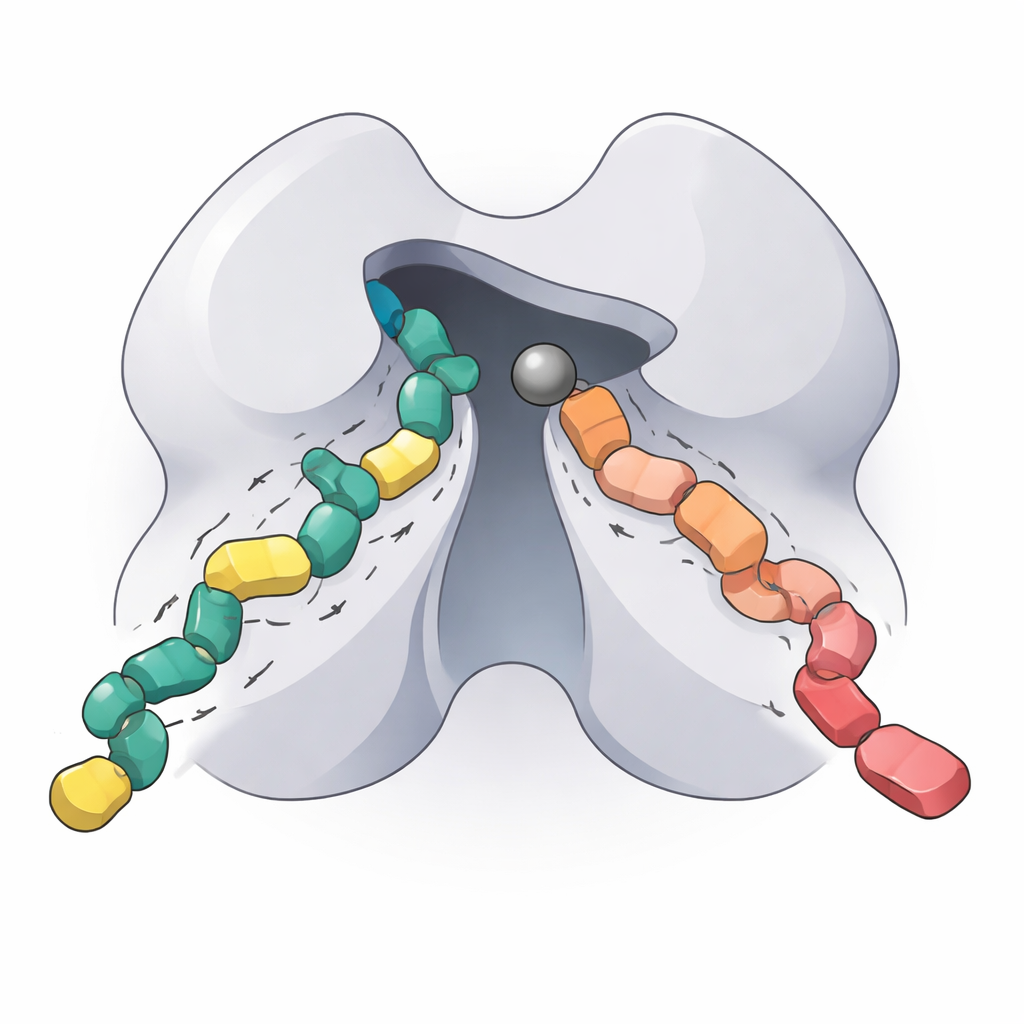
Pour combler cette lacune, les chercheurs ont produit uniquement le noyau catalytique humain d’HDAC5 — la partie qui effectue la réaction chimique — dans des bactéries, l’ont purifié et ont confirmé qu’il forme une protéine stable et monomérique en solution. Ils ont ensuite testé son fonctionnement sous différents niveaux de sel et d’acidité. L’activité d’HDAC5 est restée robuste sur une large plage de sels et atteignait un maximum en conditions légèrement basiques, comparables à celles présentes dans de nombreuses cellules. En utilisant des substrats fluorescents spéciaux, ils ont constaté que la forme naturelle d’HDAC5 reconnaît uniquement un type particulier de substrat couramment utilisé pour étudier les enzymes de la classe IIa. Guidés par des travaux antérieurs sur des HDAC apparentées, ils ont substitué un acide aminé (histidine) par une tyrosine en un point critique. De manière remarquable, ce petit changement a permis à la version mutante d’HDAC5 de traiter efficacement les deux types de substrats-test, révélant comment un seul résidu dans le site actif oriente les préférences chimiques de l’enzyme.
Tester et comparer deux nouveaux candidats-médicaments
L’équipe a ensuite examiné deux molécules expérimentales bloquant HDAC5, nommées NT160 et FFK24. Ces composés utilisent un groupe liaision au zinc plus récent qui évite une partie de la toxicité et du manque de sélectivité observés avec les anciens médicaments à base d’hydroxamate. En mesurant la façon dont chaque inhibiteur ralentissait HDAC5 dans des réactions rigoureusement contrôlées, les auteurs ont déterminé des constantes d’inhibition extrêmement faibles, de l’ordre du nanomolaire, ce qui signifie que les deux composés se lient fortement à l’enzyme. NT160 se liait de façon constante environ dix fois plus fortement que FFK24. Pour comprendre pourquoi, les chercheurs ont utilisé le docking informatique avec une structure du noyau d’HDAC5 prédite par AlphaFold. Les deux inhibiteurs partagent une zone de tête commune qui s’enfonce profondément dans la poche active et contacte l’ion métallique, mais la « queue » de NT160 formait des contacts stabilisants supplémentaires avec des acides aminés spécifiques de la poche. Ces interactions additionnelles expliquent vraisemblablement sa plus grande puissance.
Ce que cela signifie pour les thérapies ciblées à venir
En reconstituant le cœur actif fonctionnel d’HDAC5, en cartographiant ses conditions optimales de fonctionnement, en disséquant comment un changement d’un seul acide aminé modifie son comportement, et en quantifiant la liaison de deux inhibiteurs de nouvelle génération, cette étude fournit une « empreinte » biochimique détaillée d’une enzyme importante mais auparavant peu caractérisée. Pour un public non spécialiste, l’essentiel est qu’HDAC5 contribue à contrôler l’activation ou la répression de certains gènes, et que régler précisément cet interrupteur pourrait être utile pour traiter les maladies cardiaques, la neurodégénérescence, le cancer et les troubles immunitaires. Les nouvelles connaissances et les outils présentés ici devraient aider les chercheurs à concevoir des médicaments sélectifs pour HDAC5 et la classe IIa qui agissent là où c’est nécessaire tout en minimisant les effets indésirables ailleurs dans l’organisme.
Citation: Mammen, C., Hornung, F.M., Anzenhofer, C. et al. In vitro characterization of the catalytic domain of human histone deacetylase 5. Sci Rep 16, 7935 (2026). https://doi.org/10.1038/s41598-026-37633-5
Mots-clés: histone désacétylase 5, régulation épigénétique, inhibiteurs d’HDAC, thérapie ciblée contre le cancer, structure de la chromatine