Clear Sky Science · fr
Un cadre d’apprentissage profond basé sur DNABERT pour prédire les sites de liaison des facteurs de transcription
Pourquoi il est important de prédire les interrupteurs de contrôle de l’ADN
Chaque cellule de votre corps porte essentiellement le même ADN, et pourtant les cellules cérébrales, hépatiques et immunitaires se comportent très différemment. L’une des raisons est que des protéines particulières, appelées facteurs de transcription, jouent le rôle d’interrupteurs moléculaires, activant ou éteignant des gènes en se fixant sur de courts fragments d’ADN connus sous le nom de sites de liaison. Identifier expérimentalement tous ces points d’ancrage à l’échelle du génome est lent et coûteux. Cette étude présente TFBS-Finder, un nouveau modèle d’intelligence artificielle capable de lire les lettres brutes de l’ADN et de prédire plus précisément où les facteurs de transcription se lient, ce qui peut accélérer la recherche sur la régulation génétique et les maladies.
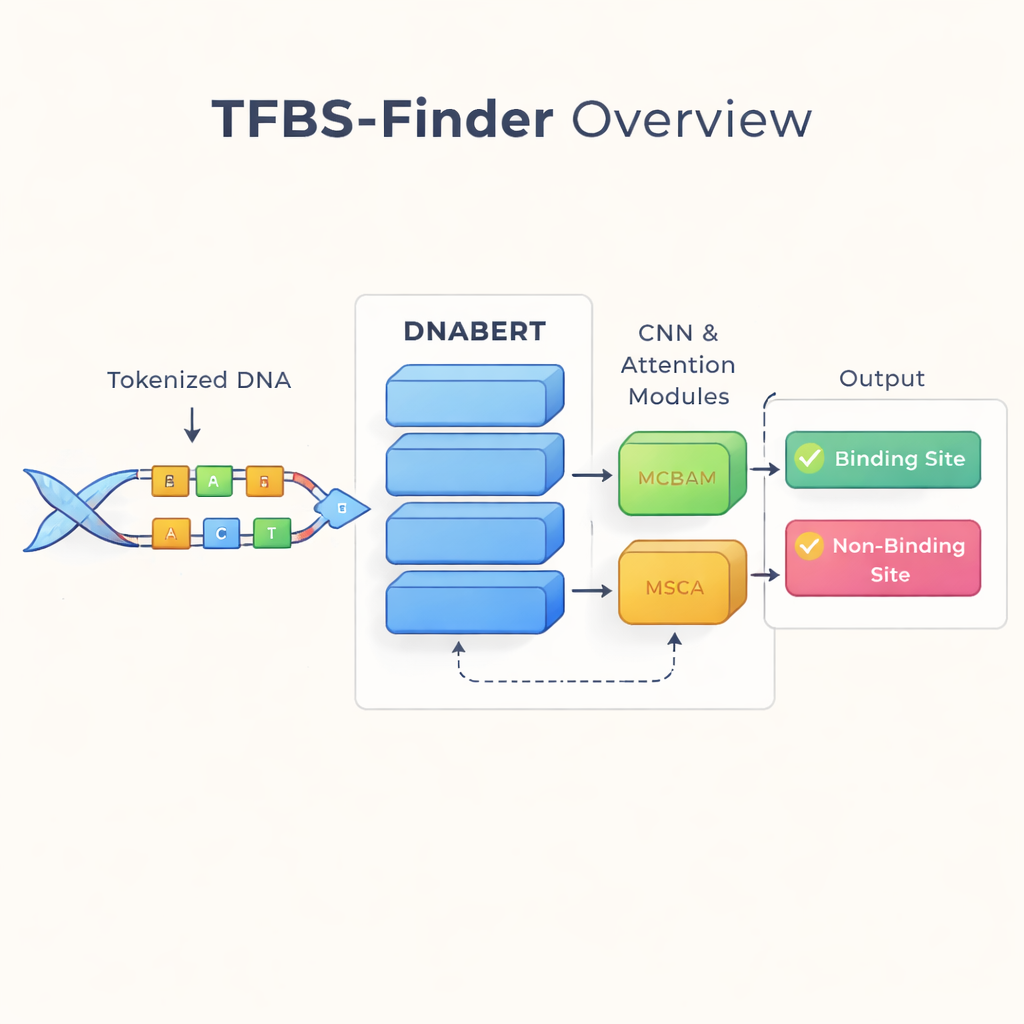
Lire l’ADN comme une langue
Les auteurs s’appuient sur une idée qui a transformé les technologies du langage : traiter l’ADN comme un texte. Ils utilisent DNABERT, une version du modèle de langage BERT réentraînée sur l’ADN humain plutôt que sur des mots. DNABERT n’examine pas les lettres isolées ; il découpe l’ADN en « mots » chevauchants de cinq lettres et apprend comment ces morceaux co‑apparaissent. Cela permet au modèle de capturer un contexte à longue portée, par exemple la façon dont des motifs à une extrémité d’une séquence se rapportent à des motifs éloignés, un peu comme comprendre le sens d’une phrase plutôt que de mots isolés.
Repérer les motifs locaux grâce à une attention ciblée
Si DNABERT saisit bien le contexte global, la liaison des facteurs de transcription dépend souvent de motifs très courts et précis — des motifs locaux dans l’ADN. TFBS-Finder ajoute donc plusieurs composants au‑dessus de DNABERT. Un réseau de neurones convolutionnel (CNN) parcourt les embeddings de séquence pour mettre en évidence des formes locales récurrentes, à la manière dont un logiciel d’image détecte des bords et des angles. Deux modules d’attention, appelés MCBAM et MSCA, fonctionnent ensuite comme des projecteurs réglables, renforçant les caractéristiques les plus informatives et atténuant le bruit. Ensemble, ces blocs équilibrent la compréhension d’ensemble et les détails fins pour décider si un segment d’ADN contient un site de liaison réel.
Montrer que chaque élément apporte vraiment quelque chose
Pour vérifier si tous ces composants sont nécessaires, l’équipe a réalisé de vastes expériences d’« ablation », en supprimant ou réorganisant systématiquement des modules et en réentraînant le système sur 165 jeux de données de référence couvrant 29 facteurs de transcription dans 32 types cellulaires. En utilisant des mesures standards de qualité de prédiction, le modèle complet TFBS-Finder est systématiquement arrivé en tête. Les versions simplifiées reposant seulement sur DNABERT, ou privées d’un des modules d’attention, ont clairement perdu en précision. Des tests statistiques ont confirmé que ces baisses de performance n’étaient pas dues au hasard, montrant que la combinaison d’une compréhension globale des séquences et d’une attention locale bien conçue est cruciale.
Fonctionner à travers les types cellulaires et battre les outils antérieurs
Une question importante est de savoir si un modèle entraîné dans un contexte biologique peut se généraliser à un autre. Les auteurs se sont concentrés sur un facteur de transcription bien étudié, CTCF, et ont entraîné TFBS-Finder sur des données d’une lignée cellulaire puis l’ont testé sur d’autres. Dans toutes les combinaisons, le modèle a obtenu des scores élevés, suggérant qu’il capture des caractéristiques essentielles de la liaison de CTCF partagées entre les tissus. Comparé à neuf méthodes de pointe, y compris d’anciens modèles d’apprentissage profond et basés sur BERT, TFBS-Finder a montré une précision moyenne supérieure et a produit des classements de sites de liaison plus fiables. Il s’est aussi exécuté légèrement plus vite et a utilisé moins de mémoire que le modèle le plus similaire précédent, indiquant qu’une meilleure performance n’exigeait pas une charge de calcul plus lourde.
Voir ce que le modèle a appris
Les systèmes d’IA complexes sont souvent critiqués comme des « boîtes noires ». Ici, les chercheurs ont tenté d’ouvrir cette boîte en visualisant quelles positions de l’ADN influençaient le plus les décisions de TFBS-Finder. Pour deux facteurs de transcription dont les motifs de liaison sont bien connus, CEBPB et GATA3, ils ont généré des scores d’importance le long de la séquence et regroupé les signaux les plus forts en motifs consensuels. Ces motifs récupérés correspondaient étroitement aux motifs de référence des bases de données établies, et les régions de liaison prédites chevauchaient des instances de motifs détectées indépendamment. Cela suggère que TFBS-Finder n’est pas simplement en train de mémoriser des exemples, mais a appris des règles biologiquement significatives sur la façon dont les facteurs de transcription reconnaissent l’ADN.
Ce que cela signifie pour la génétique et la médecine
TFBS-Finder fournit une façon plus précise et interprétable de cartographier les interrupteurs de contrôle intégrés dans notre ADN. En pinpointant les endroits où les facteurs de transcription sont susceptibles de se lier, il peut aider les chercheurs à cartographier les réseaux de régulation des gènes, à prioriser les variantes génétiques susceptibles de perturber des sites de contrôle cruciaux et à concevoir des expériences plus ciblées. Bien que le travail actuel utilise des séquences mélangées comme négatifs artificiels et se concentre uniquement sur les lettres de l’ADN, les auteurs prévoient d’ajouter des informations structurelles sur la forme de l’ADN et d’explorer des séquences de fond plus réalistes. À mesure que ces modèles s’amélioreront, ils pourraient devenir des aides puissantes pour comprendre comment les changements dans l’ADN non codant contribuent au développement, à l’évolution et au risque de maladie.
Citation: Dutta, P., Ghosh, N. & Santoni, D. A DNABERT based deep learning framework for predicting transcription factor binding sites. Sci Rep 16, 7018 (2026). https://doi.org/10.1038/s41598-026-37483-1
Mots-clés: sites de liaison des facteurs de transcription, apprentissage profond, DNABERT, régulation des gènes, génomique