Clear Sky Science · fr
Mutation GJB2 c.109G > A activant la voie d’apoptose mitochondriale médiée par IFI27 conduisant à une surdité héréditaire non syndromique
Pourquoi de minuscules cellules de l’oreille comptent pour l’avenir des enfants
La perte auditive présente à la naissance touche des millions d’enfants dans le monde et influence souvent la façon dont ils apprennent à parler, réussissent à l’école et se lient aux autres. Un des coupables génétiques les plus fréquents est un gène appelé GJB2, mais les cliniciens n’ont pas entièrement saisi comment ses altérations endommagent l’oreille interne. Cette étude utilise des poissons-zèbres et des cellules humaines pour retracer la chaîne d’événements partant d’un seul changement d’ADN dans GJB2 jusqu’à la mort de cellules sensibles au son, et identifie une nouvelle molécule, IFI27, comme cible potentielle pour de futurs traitements.
Une modification génétique fréquente derrière des silences infantiles
Les chercheurs ont commencé par dépister des échantillons sanguins de 1 199 enfants présentant une suspicion de surdité héréditaire dans la province du Fujian, en Chine. Ils se sont concentrés sur plusieurs gènes de surdité bien connus et ont constaté que les altérations de GJB2 dominaient, représentant 85 % de toutes les mutations détectées. Parmi elles, une modification spécifique appelée c.109G>A (également connue sous le nom p.Val37Ile) était la plus fréquente. Ce variant est relativement courant dans la population générale mais fortement enrichi chez les personnes présentant une perte auditive, ce qui suggère qu’il joue un rôle majeur dans la surdité non syndromique — des troubles auditifs qui surviennent sans autres problèmes médicaux.
Suivre les dommages dans un poisson transparent
Pour observer ce que cette mutation provoque dans un organisme vivant, l’équipe s’est tournée vers le poisson-zèbre, un petit poisson d’eau douce dont les embryons sont transparents et qui partage de nombreux gènes et structures de l’oreille avec l’humain. Ils ont modifié des embryons de poisson-zèbre pour exprimer soit le GJB2 humain normal, soit la version mutée c.109G>A, et ont aussi utilisé une approche d’« knockdown » pour réduire l’expression du gjb2 propre au poisson. Les embryons porteurs du gène mutant ou d’une expression réduite présentaient un retard de croissance, une queue courbée et un gonflement autour du cœur, signes d’un développement perturbé. Surtout, leurs oreilles internes étaient clairement anormales : des structures clés appelées otolithes étaient plus petites et plus éloignées, et la région cochléaire remplie de liquide était réduite. Lorsque les scientifiques ont réintroduit le GJB2 normal en même temps que le mutant, beaucoup de ces anomalies structurelles se sont améliorées, montrant que la mutation elle-même était à l’origine des défauts.
Des oreilles défaillantes à un comportement auditif altéré
Comme l’audition dépend de minuscules « cellules ciliées » qui transforment les vibrations sonores en signaux nerveux, l’équipe a coloré ces cellules chez le poisson-zèbre. Les poissons porteurs de la mutation GJB2 ou du knockdown avaient beaucoup moins de cellules ciliées tant dans l’oreille interne que le long de la surface corporelle, où le poisson-zèbre détecte aussi les mouvements de l’eau. Les chercheurs ont ensuite testé la capacité des poissons à réagir au son. À l’aide d’un système de suivi automatisé, ils ont mesuré la distance et la vitesse de nage de larves de 5 jours exposées à de brèves salves sonores. Les poissons normaux et ceux exprimant le GJB2 sauvage réagissaient en nageant plus loin et plus vite, tandis que les poissons mutants et knockdown changeaient à peine de comportement, indiquant une audition altérée. Là encore, l’ajout de GJB2 normal a partiellement restauré à la fois le nombre de cellules ciliées et les mouvements induits par le son.
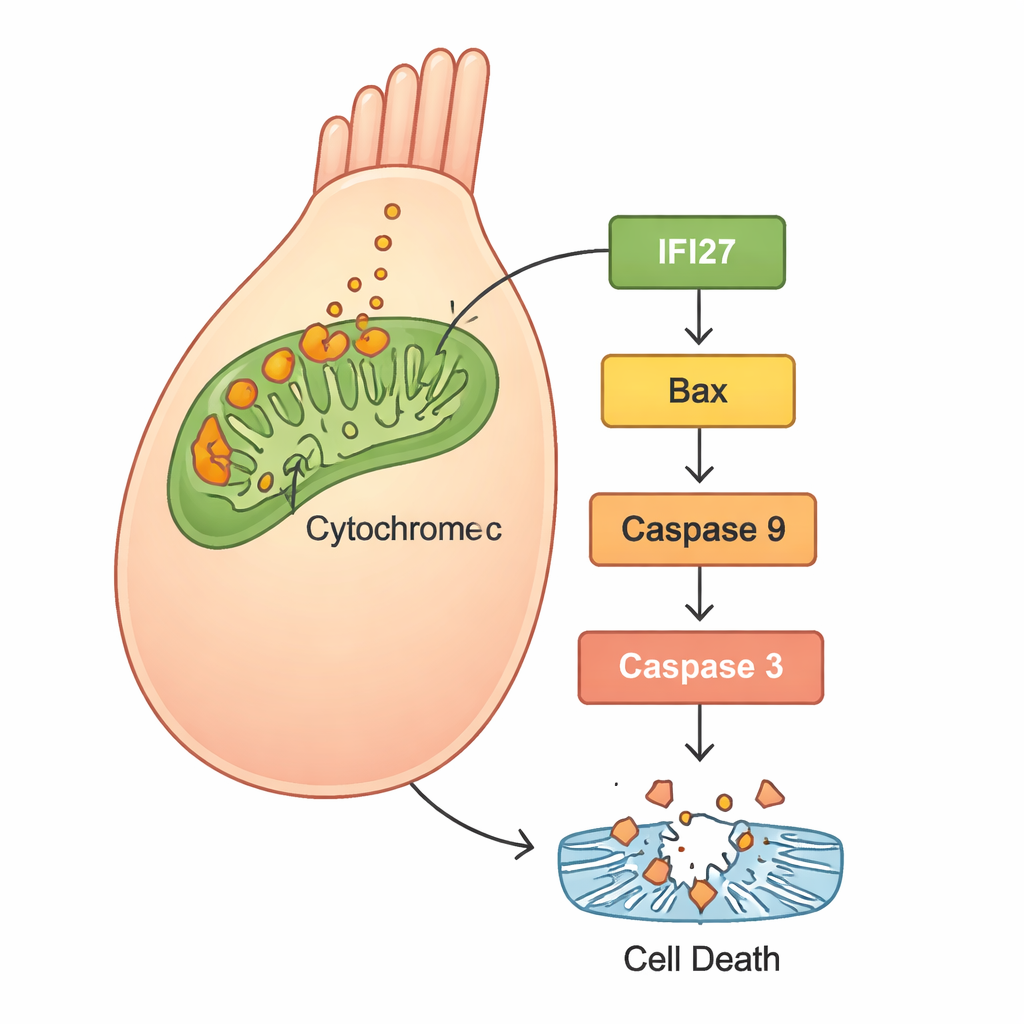
Une voie létale à l’intérieur des centrales énergétiques cellulaires
Pour comprendre ce qui se passait à l’intérieur des cellules, les chercheurs ont utilisé le séquençage de l’ARN pour comparer l’activité génique entre des poissons-zèbres normaux et ceux avec un gjb2 réduit. Un ensemble de gènes lié à la « voie d’apoptose mitochondriale » — une voie d’autodestruction centrée dans les centrales énergétiques de la cellule — était fortement activé. En particulier, plusieurs membres de la famille IFI27 se distinguaient, ainsi que des acteurs connus de la mort cellulaire comme Bax, le cytochrome c, Apaf1 et des caspases. Des expériences complémentaires dans des cellules humaines HEK293 ont confirmé le schéma : les cellules portant le GJB2 mutant produisaient davantage d’espèces réactives de l’oxygène (ROS, une forme de stress oxydatif), libéraient plus de cytochrome c des mitochondries et activaient des protéines d’apoptose, conduisant à une augmentation de la mortalité cellulaire. Lorsque les chercheurs ont réduit l’expression d’IFI27 dans des cellules portant le gène mutant, les niveaux de ROS ont diminué, les signaux de mort ont été atténués et moins de cellules ont subi l’apoptose.
Ce que cela implique pour de futurs traitements
Dans l’ensemble, les résultats dessinent un scénario clair : la mutation GJB2 c.109G>A perturbe le développement et la fonction de l’oreille interne, non seulement en altérant la communication cellulaire, mais aussi en déclenchant un stress mitochondrial. Ce stress augmente l’expression d’IFI27 et de gènes associés, libère le cytochrome c et déclenche une cascade de protéines poussant les cellules ciliées vers la mort programmée. Comme les cellules ciliées ne repoussent pas facilement chez l’humain, leur perte entraîne des déficits auditifs permanents. En montrant que la diminution d’IFI27 peut atténuer cette cascade destructrice dans des cellules humaines, l’étude met en avant IFI27 comme une cible prometteuse pour des médicaments ou des thérapies géniques. Si de tels traitements restent lointains — et nécessiteront probablement d’être administrés très tôt dans la vie — ce travail fournit une feuille de route moléculaire concrète pour transformer une mutation autrefois mystérieuse en une cause potentiellement évitable de surdité infantile.
Citation: Chen, Y., Zhao, P., Lin, Q. et al. GJB2 c.109G > A mutation activating IFI27-mediated mitochondrial apoptosis pathway leading to hereditary non-syndromic hearing loss. Sci Rep 16, 6240 (2026). https://doi.org/10.1038/s41598-026-37393-2
Mots-clés: surdité génétique, mutation GJB2, modèle poisson-zèbre, apoptose mitochondriale, IFI27