Clear Sky Science · fr
Un cadre tri-omique et d’apprentissage automatique identifie des biomarqueurs pronostiques et des signatures métaboliques dans le sepsis
Pourquoi cela compte pour les personnes atteintes d’infections sévères
Le sepsis est une réaction potentiellement mortelle à une infection qui peut propulser le système immunitaire en hyperactivité et entraîner une défaillance d’organes. Les médecins savent que détecter le sepsis tôt et adapter le traitement à chaque patient peut sauver des vies, mais les tests sanguins actuels sont des outils peu fins : ils donnent souvent peu d’indications sur qui va s’améliorer et qui présente le plus de risques. Cette étude utilise une combinaison puissante de trois types de mesures moléculaires et de l’apprentissage automatique moderne pour chercher des signaux d’alerte plus précis dans le sang des patients septicémiques.
Regarder le sang à travers trois lentilles différentes
Plutôt que de se concentrer sur un seul type de molécule, les chercheurs ont profilé les mêmes patients de trois manières simultanément. Ils ont mesuré quels gènes étaient activés ou réprimés (transcriptomique), quelles protéines étaient réellement présentes et actives (protéomique), et quels petits métabolites circulaient (métabolomique). Ils ont prélevé du sang chez 21 patients atteints de sepsis et 10 volontaires sains et ont utilisé des méthodes statistiques avancées pour voir comment ces trois couches évoluaient ensemble dans la maladie. Cette vision « tri-omique » aide à surmonter un problème clé : dans le sepsis, l’activité génique et les niveaux protéiques peuvent se découpler, si bien qu’analyser une seule couche peut être trompeur.
Apprendre aux algorithmes à repérer les profils à haut risque
Parmi des milliers de gènes et de protéines, l’équipe a d’abord utilisé une méthode de réseau pour trouver des groupes qui évoluaient de concert dans le sepsis. Ils ont ensuite recoupé ces groupes avec les protéines qui différaient nettement entre patients et témoins sains, obtenant 32 candidats solides. Pour affiner encore cette liste, ils ont eu recours à l’apprentissage automatique, employant deux algorithmes complémentaires pour éliminer les signaux faibles et ne conserver que les plus informatifs. Lorsqu’ils ont testé la relation entre ces gènes restants et la survie dans une grande base de données publique sur le sepsis, deux gènes ont émergé : TPR et ERN1. Des niveaux plus élevés de TPR étaient associés à une survie plus longue, tandis que des niveaux plus élevés d’ERN1 étaient liés à de moins bons pronostics.
Relier les cellules immunitaires et le métabolisme perturbé
L’étude ne s’est pas arrêtée aux gènes et aux protéines. En analysant des milliers de métabolites dans le sang des patients, les chercheurs ont identifié 136 petites molécules qui suivaient étroitement TPR et ERN1. Nombre d’entre elles appartiennent à des voies traitant les lipides membranaires et les acides gras, essentielles à la signalisation des cellules immunitaires et à la propagation de l’inflammation. Parallèlement, une analyse en cellule unique — qui examine les cellules immunitaires individuelles plutôt que des échantillons mélangés — a montré que TPR et ERN1 sont particulièrement actifs dans les monocytes, les macrophages et les cellules NK. Ensemble, ces résultats suggèrent que ces deux marqueurs occupent une position charnière entre les cellules combattant l’infection et la façon dont ces cellules utilisent et remodelent les lipides et l’énergie pendant le sepsis.
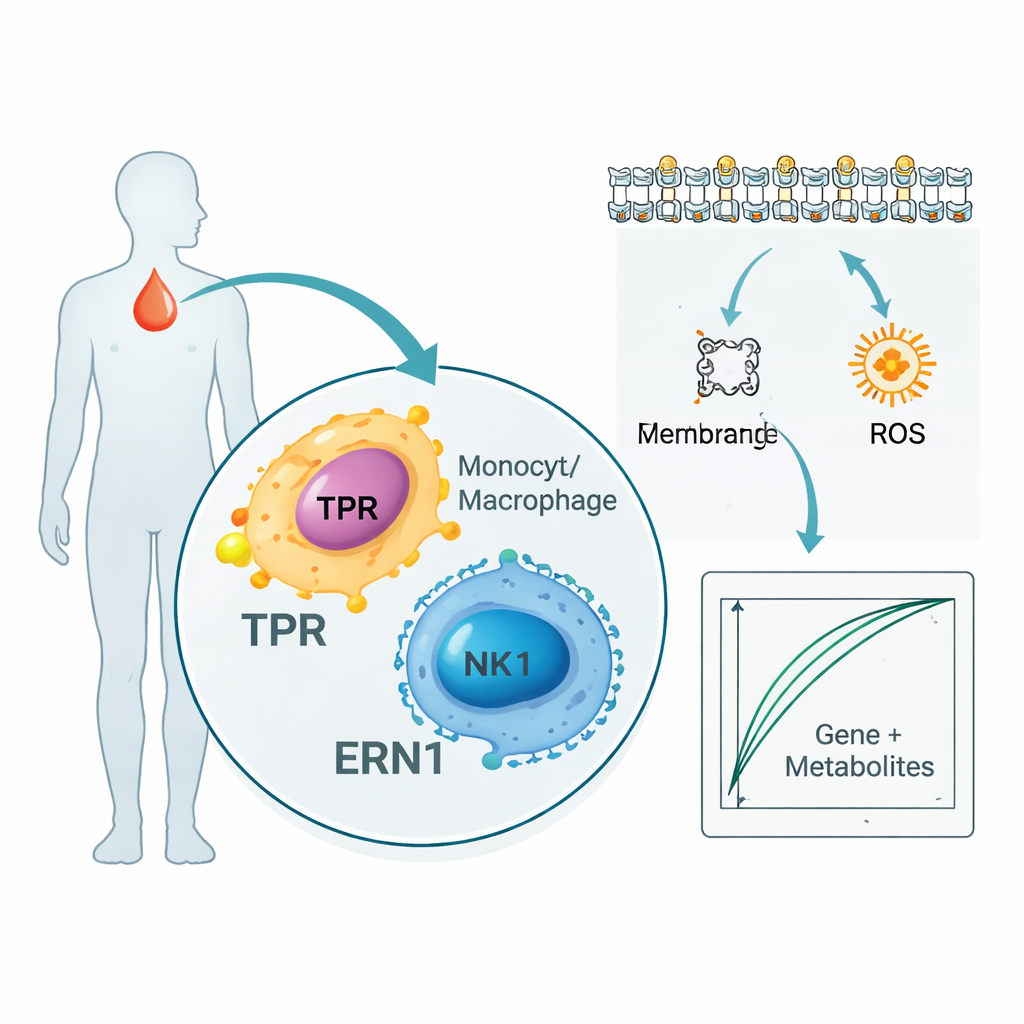
Construire un test sanguin proof‑of‑concept
Pour explorer la traduction possible de ces découvertes en pratique, les auteurs ont combiné les deux gènes avec cinq des métabolites les plus informatifs pour entraîner des modèles informatiques simples séparant les patients septicémiques des personnes saines. Dans leur petit jeu de données interne, ces signatures combinées « gène-plus-métabolite » ont presque parfaitement distingué qui avait un sepsis. Les chercheurs ont aussi consulté de grandes bases publiques qui relient les protéines sanguines au risque de maladie chez des dizaines de milliers de personnes et ont constaté que les niveaux protéiques de TPR et ERN1 étaient systématiquement associés à des affections liées au sepsis, apportant une couche supplémentaire de soutien. Néanmoins, les auteurs insistent sur le fait que ces modèles sont des outils en phase précoce destinés à générer des hypothèses, et non des tests prêts à l’emploi au chevet des patients.
Des composés végétaux comme pistes initiales, pas comme remèdes
Dans une dernière étape, l’équipe s’est demandé si des molécules naturelles pouvaient influencer TPR ou ERN1. Ils ont interrogé une base de données spécialisée de près de 500 composés purifiés issus de la médecine traditionnelle chinoise, chacun avec son propre profil d’activité génique. Plusieurs composés semblaient fortement augmenter ou diminuer ces deux gènes dans des cellules cultivées en laboratoire, suggérant qu’ils pourraient un jour aider les chercheurs à explorer la biologie du sepsis ou à concevoir de nouveaux médicaments. Cependant, ces résultats proviennent uniquement d’un appariement informatique : ils ne démontrent pas que l’une de ces substances soit sûre ou efficace chez les personnes atteintes de sepsis.
Ce que ce travail nous apprend réellement
Cette étude offre une carte détaillée plutôt qu’une solution achevée. En tissant ensemble trois couches moléculaires, des données en cellule unique et l’apprentissage automatique, les auteurs mettent en lumière TPR et ERN1 — et leurs changements métaboliques associés — comme des balises prometteuses de la façon dont le système immunitaire et le métabolisme se déséquilibrent dans le sepsis. Pour un lecteur non spécialiste, le message clé est que le sepsis n’est pas une maladie unique mais un ensemble changeant d’états immunitaires et métaboliques, et que des tests sanguins plus intelligents pourraient un jour aider les cliniciens à identifier l’état d’un patient et à adapter le traitement en conséquence. Avant d’en arriver là, ces signaux préliminaires doivent être testés et confirmés dans des cohortes beaucoup plus larges et diversifiées, ainsi que dans des expérimentations en laboratoire capables d’établir causalité.
Citation: Li, X., Ke, G., Hu, Y. et al. A tri-omics and machine learning framework identifies prognostic biomarkers and metabolic signatures in sepsis. Sci Rep 16, 6648 (2026). https://doi.org/10.1038/s41598-026-37342-z
Mots-clés: biomarqueurs du sepsis, multi-omique, apprentissage automatique en médecine, métabolisme immunitaire, diagnostics de précision