Clear Sky Science · fr
Dépistages à l’échelle du génome identifient les régulateurs centraux de l’expression de la protéine prion à la surface cellulaire
Pourquoi c’est important pour la santé cérébrale
Les maladies à prions, comme la maladie de Creutzfeldt–Jakob chez l’humain et l’« encéphalopathie spongiforme bovine » chez le bovin, sont rares mais toujours fatales. L’un des acteurs centraux est une protéine cérébrale normale, appelée protéine prion, qui peut se replier de manière pathologique et propager des lésions de cellule en cellule. Plus cette protéine est présente à la surface des neurones, plus la maladie peut s’installer facilement. Cette étude a cherché à cartographier, à l’échelle du génome, les gènes qui contrôlent la quantité de protéine prion affichée à l’extérieur des cellules de type neuronal. Cette cartographie pourrait aider les chercheurs à concevoir des moyens de réduire cette protéine et, potentiellement, de ralentir plusieurs maladies neurodégénératives.
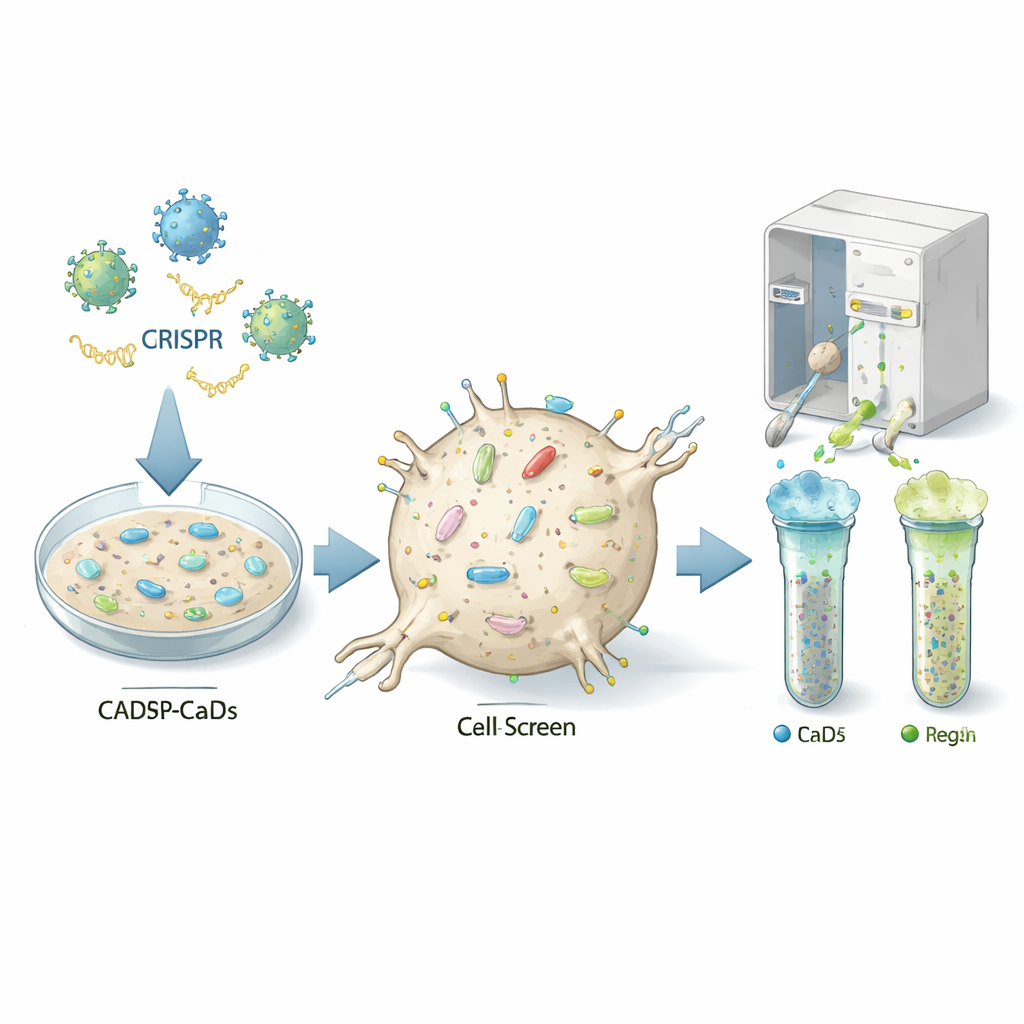
Trouver les boutons de commande de la cellule
Les auteurs ont utilisé une puissante méthode d’édition génétique, le CRISPR, pour inactiver presque chaque gène, un par un, dans une lignée cellulaire de souris de type neuronal susceptible à l’infection par les prions (appelée cellules CAD5). Chaque cellule a reçu un « coup » génétique différent, de sorte que la population résultante contenait des millions de variantes, chacune privée d’un gène spécifique. L’équipe a ensuite marqué les cellules avec des anticorps fluorescents reconnaissant la protéine prion normale à la surface cellulaire et utilisé un trieur cellulaire pour séparer les cellules présentant des niveaux inhabituellement bas ou élevés de cette protéine. En séquençant les ARN guides enrichis dans les groupes faibles ou forts, ils ont pu déduire quels gènes inactivés agissent normalement comme interrupteurs marche/arrêt pour la protéine prion à la surface cellulaire.
Deux états cellulaires, des réponses qui se recoupent
Les neurones ne sont pas tous identiques au cours de leur vie : leur apparence et leur comportement changent. Les chercheurs ont donc vérifié si les mêmes gènes contrôlent la protéine prion selon l’état cellulaire. Les cellules CAD5 peuvent être maintenues dans un état prolifératif, moins spécialisé, ou être amenées, par retrait du sérum du milieu, à adopter une forme plus mature et neuronale. L’équipe a réalisé le même crible CRISPR à l’échelle du génome dans les deux conditions. Dans les cellules indifférenciées (moins matures), ils ont validé 46 gènes qui augmentent et 21 qui diminuent la présence de la protéine prion à la surface lorsqu’ils sont présents. Dans les cellules différenciées (plus neuronales), ils ont confirmé 41 régulateurs positifs et 13 régulateurs négatifs. Vingt-trois gènes — principalement ceux impliqués dans l’attachement d’une « ancre » lipidique à la protéine — étaient communs aux deux états cellulaires, mettant en évidence un noyau de régulation opérant indépendamment de la maturité.
Lignes d’assemblage clés qui importent le plus
Une analyse plus approfondie a révélé que nombre des gènes nouvellement identifiés appartiennent à des « lignes d’assemblage » cellulaires connues qui modifient les protéines au fur et à mesure de leur trajet vers la surface. Une voie majeure assemble l’ancre GPI, une petite structure riche en lipides qui attache la protéine prion à la face externe de la membrane cellulaire. L’altération de presque n’importe quelle étape de cette voie a réduit la quantité de protéine prion atteignant la surface, tant dans les cellules immatures que matures. Une deuxième voie implique la N-glycosylation, au cours de laquelle des chaînes de sucres complexes sont ajoutées aux protéines lors de leur passage dans les membranes internes de la cellule. Les gènes de cette voie d’ajout de sucres se sont révélés principalement importants dans les cellules moins matures. Lorsque les chercheurs ont traité les cellules par de petites molécules bloquant des étapes spécifiques de la glycosylation, les niveaux de protéine prion à la surface ont diminué d’environ un tiers sans tuer les cellules, corroborant les résultats génétiques.
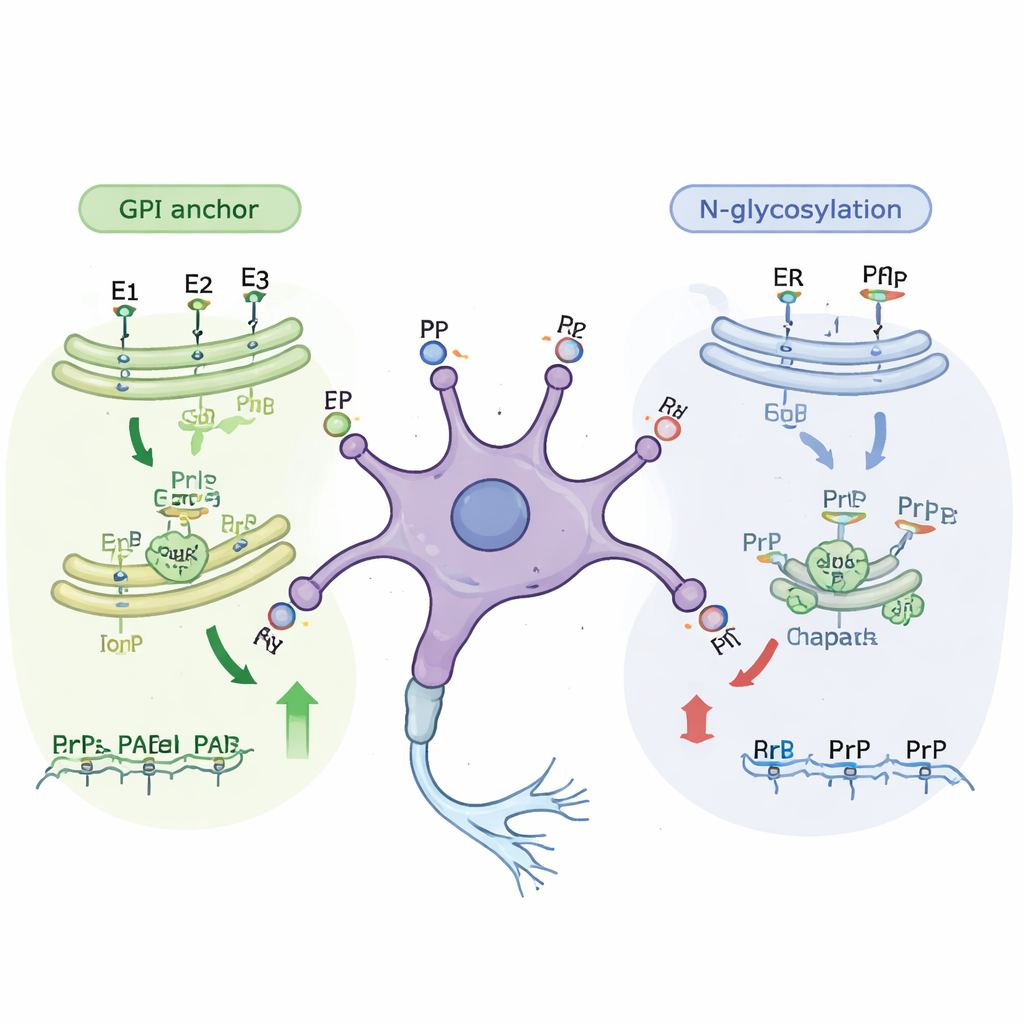
Protéines aides et réponses au stress
Les cribles ont également mis en lumière des chaperonnes moléculaires — des protéines qui aident d’autres protéines à se replier correctement — comme régulateurs importants de la protéine prion. En particulier Hspa5 (aussi appelée BiP), une chaperonne centrale du compartiment de repliement des protéines, est apparue comme régulateur positif dans les cellules plus neuronales. Lorsque les chercheurs ont utilisé un médicament pour inhiber Hspa5, les niveaux de protéine prion à la surface ont diminué dans les deux états cellulaires, là encore sans dommage évident pour les cellules. D’autres gènes identifiés sont impliqués dans le transport des protéines à travers la cellule, le contrôle de l’activation génique, ainsi que plusieurs protéines liées à la fonction synaptique et à d’autres maladies cérébrales comme la maladie d’Alzheimer et la SLA. Ensemble, ces résultats montrent que les niveaux de protéine prion à la surface sont façonnés par un réseau de voies couvrant la production, la modification, le trafic et le contrôle qualité des protéines.
Ce que cela signifie pour les traitements futurs
Ce travail fournit le premier catalogue exhaustif des gènes qui contrôlent la quantité de protéine prion affichée à la surface de cellules de type neuronal susceptibles à l’infection par les prions. Certains de ces gènes, en particulier ceux des voies d’ancrage GPI et de N-glycosylation et le système de chaperonnes Hspa5, apparaissent comme des points de départ prometteurs pour la découverte de médicaments : réduire leur activité devrait diminuer la quantité de protéine prion disponible pour se replier de façon pathologique, et des études antérieures montrent que même des réductions partielles peuvent retarder significativement la maladie chez l’animal. En parallèle, les différences nettes entre cellules immatures et matures soulignent que l’état des cellules cérébrales importe lors du choix de cibles. Bien que davantage de travaux soient nécessaires pour tester comment la manipulation de ces gènes affecte l’infection à prions et d’autres pathologies neurodégénératives in vivo, cette étude offre une feuille de route des leviers cellulaires que les chercheurs peuvent explorer pour ralentir ou prévenir ces maladies dévastatrices.
Citation: Beauchemin, K.S., Supattapone, S. Genome-wide screens identify core regulators of cell surface prion protein expression. Sci Rep 16, 5895 (2026). https://doi.org/10.1038/s41598-026-37137-2
Mots-clés: protéine prion, crible CRISPR, neurodégénérescence, glycosylation des protéines, ancre GPI