Clear Sky Science · fr
Sélection de caractéristiques simple, rapide et efficace guidée par un regroupement flou adaptatif pour données microarray biomédicales binaires à haute dimension et fortement déséquilibrées
Pourquoi cela compte pour la recherche génétique
Les tests modernes d’expression génique peuvent mesurer des dizaines de milliers de gènes sur un seul échantillon patient. Cet afflux de données promet un diagnostic du cancer plus précoce et de meilleurs choix thérapeutiques, mais il crée aussi un problème : la plupart de ces gènes sont bruyants, redondants, ou liés principalement aux cas fréquents plutôt qu’aux cas rares et dangereux. Cet article présente une nouvelle méthode pour trier d’immenses jeux de données d’expression génique afin que les ordinateurs puissent repérer de manière fiable les patients appartenant à une petite minorité difficile à détecter en n’utilisant qu’un ensemble de gènes très réduit et soigneusement choisi.
Le problème du trop grand nombre de gènes trop similaires
Les expérimentations sur microarray suivent souvent des milliers de niveaux d’activité génique pour seulement quelques centaines de patients. Le plus souvent, une classe (par exemple un sous‑type de cancer courant) surreprésente largement l’autre, créant des données fortement déséquilibrées. Dans ce contexte, de nombreux gènes se comportent de façon très similaire et les profils des patients majoritaires et minoritaires peuvent se chevaucher. Les méthodes d’apprentissage classiques ont tendance à s’accrocher à la classe majoritaire et à être perturbées par des gènes redondants, ce qui conduit à du surapprentissage et à une mauvaise détection des sous‑types rares. Les méthodes traditionnelles de réduction de dimension perdent soit en interprétabilité en construisant de nouvelles caractéristiques mixtes, soit sélectionnent des gènes sans examiner de près dans quelle mesure ils aident un classifieur à reconnaître les cas minoritaires.
Une nouvelle feuille de route pour une sélection de gènes plus intelligente
Les auteurs introduisent AFCG‑SFE, un modèle de sélection de caractéristiques adaptatif conçu spécifiquement pour les données d’expression génique à haute dimension et déséquilibrées. La méthode démarre par une recherche simple de type « agent unique » qui active ou désactive des gènes et teste leur utilité pour la classification, mais l’enrichit par plusieurs étapes pilotées par les données. D’abord, elle regroupe les gènes selon leurs comportements similaires, puis autorise l’appartenance multiple d’un gène à plusieurs groupes pour refléter la réalité biologique qu’un gène peut participer à plusieurs voies. Au sein de chaque groupe, elle classe les gènes selon leur information vis‑à‑vis de l’étiquette de la maladie et ne conserve que quelques représentants clés, réduisant fortement la redondance avant même le début de la recherche principale. 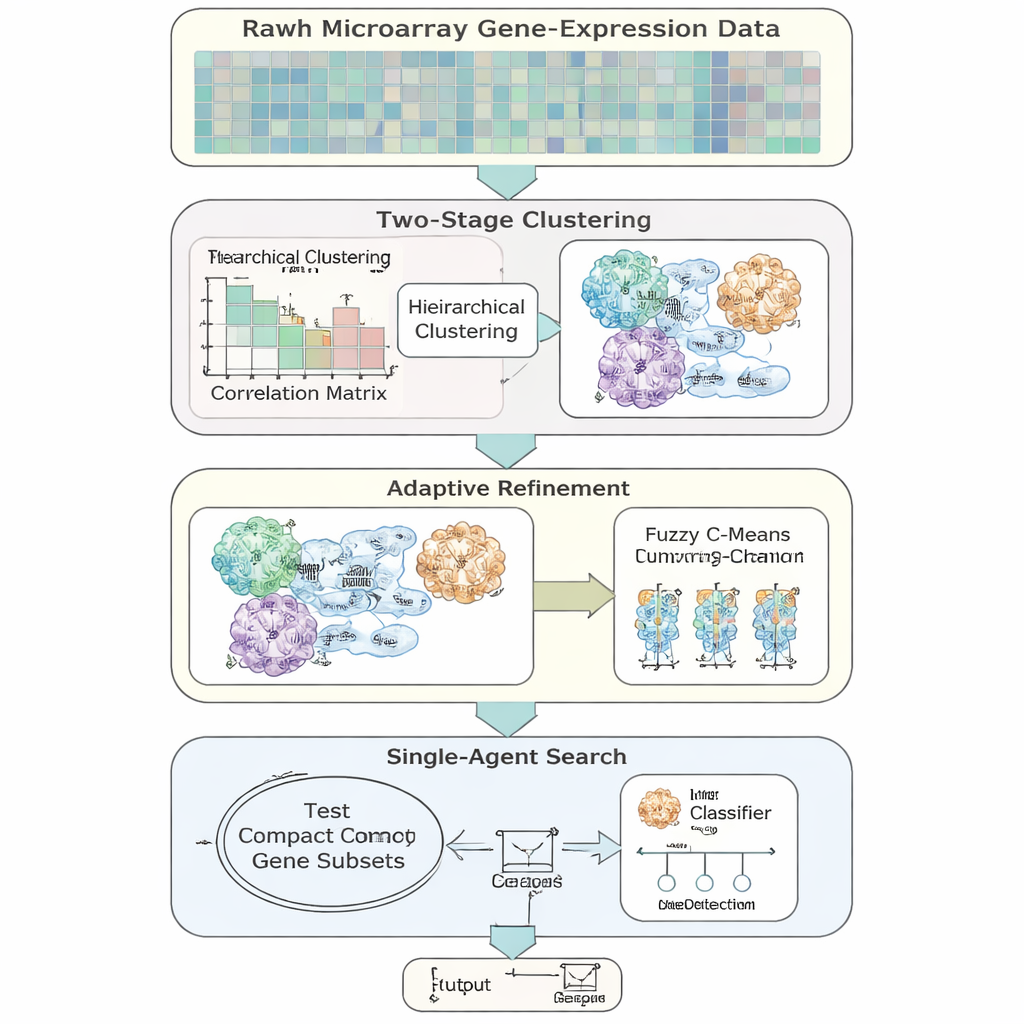
Faire en sorte que l’ordinateur s’intéresse aux patients rares
Plutôt que de se focaliser sur la seule exactitude globale, AFCG‑SFE utilise un score d’aptitude qui met l’accent sur des métriques adaptées aux données déséquilibrées, incluant l’équilibre entre l’identification correcte des cas minoritaires et majoritaires et la performance sur l’ensemble des seuils de décision. La fonction d’aptitude inclut aussi des pénalités pour la sélection d’un nombre excessif de gènes ou de nombreux gènes issus du même cluster, et une récompense pour les gènes présentant une forte dépendance vis‑à‑vis de l’étiquette de la maladie. Fait important, l’intensité de ces pénalités et récompenses est déterminée automatiquement à partir des propriétés du jeu de données, telles que le nombre de gènes par patient et le degré de chevauchement des classes, plutôt que par un réglage manuel. Cela rend la méthode plus robuste et plus facile à transférer d’une étude à l’autre.
S’adapter à la difficulté du problème
Une idée clé est que l’algorithme ne doit pas systématiquement viser l’ensemble de gènes le plus petit possible. Lorsque les deux classes sont très difficiles à séparer ou fortement chevauchées, la méthode augmente automatiquement une borne inférieure sur le nombre de gènes à conserver, garantissant que des signaux rares mais importants ne soient pas écartés. Au fur et à mesure de la recherche, AFCG‑SFE resserre progressivement un plafond par cluster sur le nombre de gènes pouvant survivre dans chaque groupe, tout en respectant ce minimum. Le résultat est un panel compact et diversifié de gènes qui capture la structure des données sans être dominé par un motif unique et redondant. 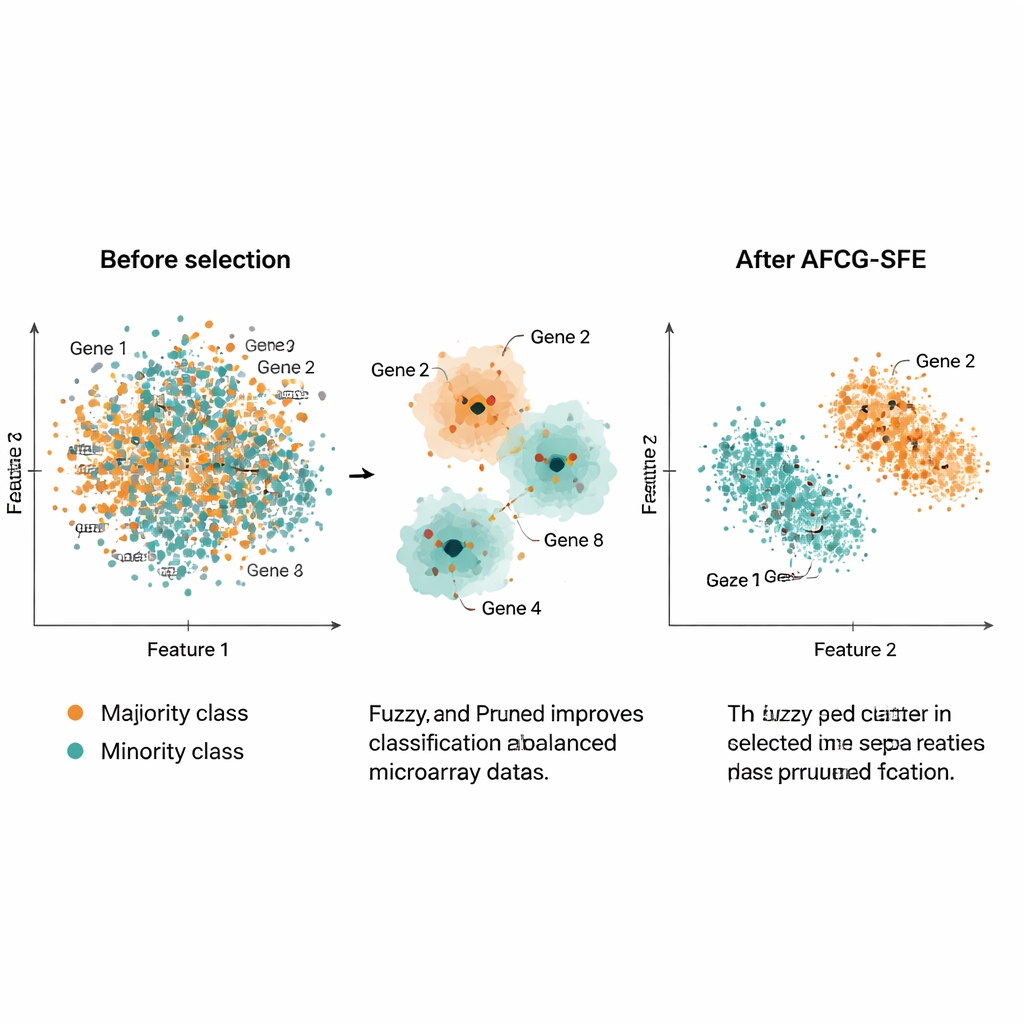
Ce que montrent les expériences
Les auteurs ont testé AFCG‑SFE sur 20 jeux de données microarray publics de cancers, chacun comportant des milliers de gènes mais seulement environ 100–200 échantillons et un fort déséquilibre de classes. Ils ont comparé leur méthode à plusieurs bases de recherche évolutionnaire, filtres simples et approches intégrées qui incorporent la sélection de caractéristiques dans le classifieur. Sur une batterie de mesures — y compris la F‑mesure, l’exactitude équilibrée, l’aire sous la courbe ROC et une mesure du surapprentissage — AFCG‑SFE a été la meilleure ou ex æquo sur tous les jeux de données. Elle sélectionnait typiquement moins de 25 gènes (souvent aussi peu que 6–8), éliminant plus de 99 % des caractéristiques initiales tout en améliorant ou en maintenant la performance de classification. Elle a également réduit un indice de complexité capturant le degré de chevauchement des classes dans l’espace des caractéristiques, indiquant une séparation plus nette après la sélection.
Conclusion pour les non‑spécialistes
Concrètement, ce travail propose une façon de réduire d’énormes profils d’expression génique bruyants à des ensembles très petits de gènes informatifs permettant néanmoins aux modèles de reconnaître avec précision des sous‑groupes de patients rares. En regroupant intelligemment les gènes similaires, en récompensant ceux qui suivent véritablement la maladie et en se prémunissant explicitement contre le biais en faveur de la classe majoritaire, AFCG‑SFE offre à la fois de meilleures prédictions et des panels de gènes beaucoup plus simples. Cette combinaison peut aider les chercheurs à identifier des biomarqueurs potentiels, concevoir des tests diagnostiques plus interprétables et, en fin de compte, améliorer la manière dont les outils de médecine de précision fonctionnent avec des données biologiques réelles et imparfaites.
Citation: Tye, Y.W., Chew, X., Yusof, U.K. et al. Adaptive fuzzy cluster-guided simple, fast, and efficient feature selection for high-dimensional and highly imbalanced binary-class bioinformatics microarray data. Sci Rep 16, 6650 (2026). https://doi.org/10.1038/s41598-026-37086-w
Mots-clés: expression génique, sélection de caractéristiques, données déséquilibrées, microarray, sous‑types de cancer