Clear Sky Science · fr
rhinotypeR permet une attribution reproductible des génotypes de rhinovirus à partir de séquences VP4/2
Pourquoi les petits virus du rhume comptent encore
La plupart d’entre nous considèrent le rhume comme une gêne plutôt que comme une menace sérieuse. Pourtant, les virus responsables de nombreux rhumes — les rhinovirus humains — sont aussi associés à des infections pulmonaires sévères, des crises d’asthme et des exacerbations de maladies respiratoires chroniques. Pour suivre l’évolution et la diffusion de ces virus, les chercheurs doivent les classer en « types » génétiques précis, un peu comme attribuer des codes‑barres aux produits. Cet article présente rhinotypeR, un logiciel libre et open‑source qui rend cette étiquetage génétique plus précis, cohérent et reproductible, aidant ainsi les équipes de santé publique à mieux surveiller une famille de virus respiratoires souvent négligée.
La diversité cachée des rhumes courants
Les rhinovirus humains sont extraordinairement répandus, apparaissant dans jusqu’à 60 % des prélèvements chez des personnes présentant des symptômes respiratoires aigus. Loin d’être un seul virus, ils sont divisés en trois groupes principaux, appelés A, B et C, et au moins 169 types génétiques reconnus. Les différents types se comportent différemment : certains sont plus souvent liés à des infections sévères chez les enfants et à des poussées d’asthme, tandis que d’autres sont moins fréquemment observés dans les formes graves. Parce que ces types évoluent indépendamment et présentent des caractéristiques de surface distinctes, les chercheurs ont besoin de méthodes fiables pour les distinguer s’ils veulent suivre la propagation des épidémies dans les écoles, les foyers et les communautés.
De outils épars à une voie unique
Jusqu’à présent, déterminer le type d’un rhinovirus à partir de son code génétique relevait d’un travail morcelé. Les chercheurs se concentraient généralement sur un court segment du génome viral appelé région VP4/2, l’alignaient sur des souches de référence connues, mesuraient les distances entre les séquences, puis appliquaient des seuils pour décider du type de chaque échantillon. Mais ces étapes étaient réalisées avec un mélange de programmes logiciels, de corrections manuelles et de jugements personnels. Cela rendait difficile la comparaison ou la reproductibilité des études, même avec des données similaires. rhinotypeR a été créé spécifiquement pour transformer ce processus en plusieurs étapes, sujet aux erreurs, en un flux de travail scripté unique que chacun peut exécuter et partager.
Ce que fait réellement le nouveau logiciel
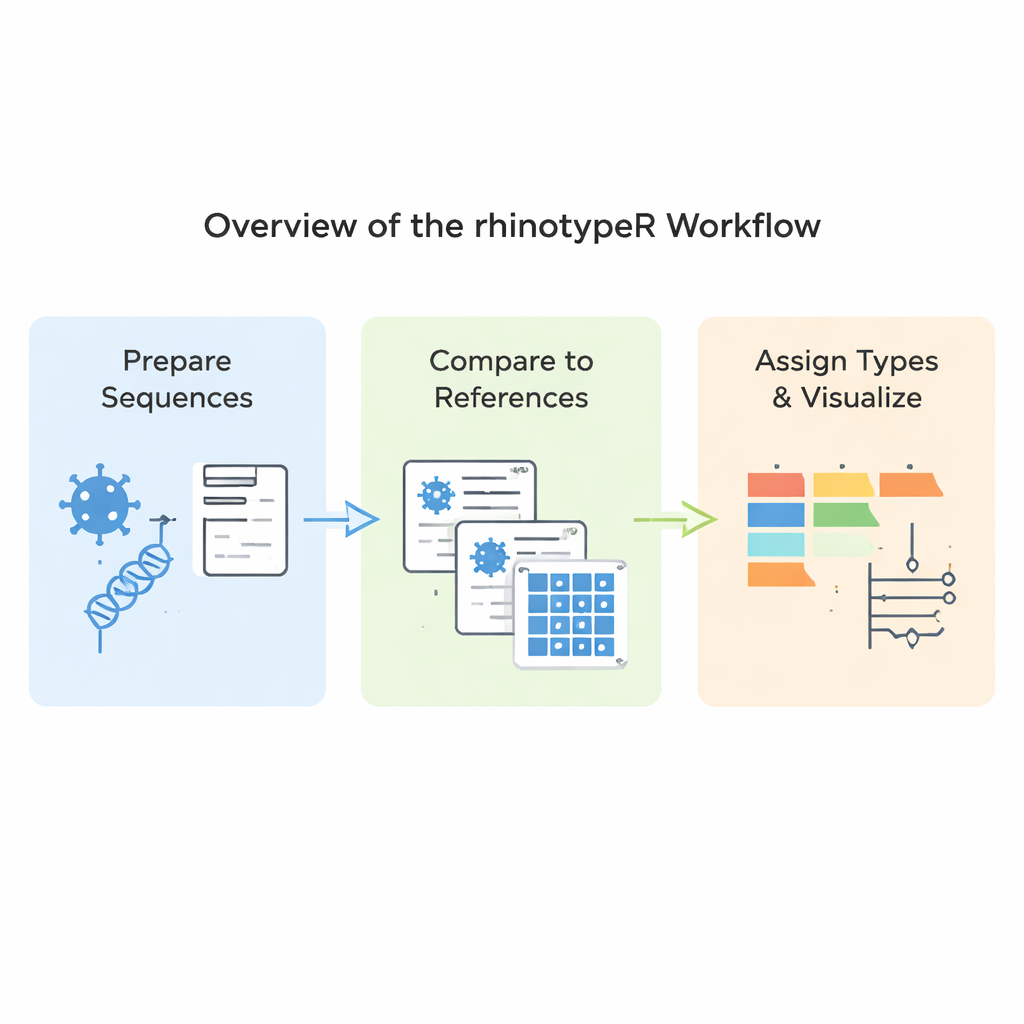
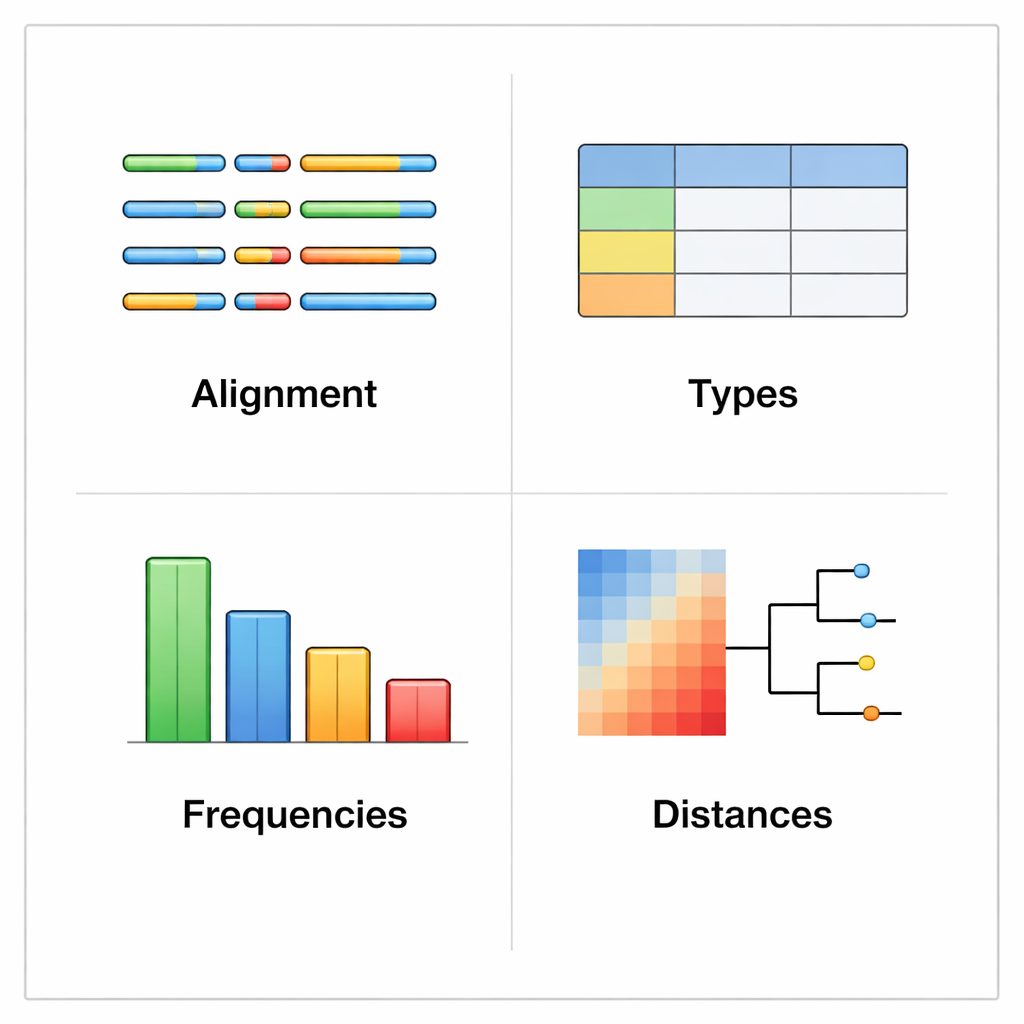
rhinotypeR fonctionne dans l’environnement R et Bioconductor, largement utilisé pour l’analyse de données. Il prend en entrée un ensemble de séquences VP4/2 de rhinovirus et les conduit à travers trois étapes principales : préparer et aligner les séquences, calculer la distance de chacune par rapport à un ensemble de types de référence curés, puis attribuer chaque échantillon au type connu le plus proche ou le signaler comme « non attribué » s’il est trop différent. Le même outil peut produire des sorties visuelles, notamment des cartes colorées des différences génétiques, des arbres de parenté simplifiés et des graphiques montrant la prévalence de chaque type dans un jeu de données. Les utilisateurs peuvent aligner leurs données avec des programmes externes s’ils le préfèrent, ou laisser rhinotypeR gérer l’intégralité du processus dans R pour une reproductibilité maximale.
Mettre l’outil à l’épreuve
Pour vérifier que rhinotypeR fournit des résultats fiables, les auteurs ont comparé ses mesures de distance à celles de deux programmes établis, ape et MEGA X, en utilisant les mêmes fichiers d’entrée et modèles. Les résultats concordent presque parfaitement ; les petites différences observées tenaient à l’arrondissement normal en calcul informatique, et non à des divergences méthodologiques réelles. L’équipe a ensuite appliqué rhinotypeR à une large collection de plus de 2 300 séquences de rhinovirus issues d’études antérieures, couvrant plus de 90 % des types connus. Dans environ quatre cas sur cinq, le nouvel outil était en accord exact avec les étiquettes de type précédentes. La plupart des désaccords se situaient juste autour des seuils préalablement convenus utilisés pour séparer un type d’un autre, précisément là où les appels limites sont attendus. Fait important, les échantillons qui n’ont pas pu être attribués avec confiance à un type connu ne présentaient pas de signes d’une mauvaise qualité ou d’une faible charge virale, ce qui suggère qu’ils reflètent peut‑être une véritable diversité virale.
Pourquoi cela compte pour la santé publique
Pour les non‑spécialistes, l’essentiel est que rhinotypeR ne réinvente pas la classification des virus du rhume ; il rend plutôt ce processus plus clair, plus transparent et plus reproductible. En regroupant l’alignement, le calcul des distances et l’attribution des types dans un seul package open‑source — accompagné de résumés visuels clairs — il aide les chercheurs et les programmes de surveillance à traiter des milliers d’échantillons de manière cohérente. Cette cohérence améliore notre capacité à comparer des études menées en différents lieux et à différentes époques, à détecter tôt des lignées virales inhabituelles ou émergentes, et à relier des motifs génétiques à des tendances épidémiologiques réelles. À long terme, des outils comme rhinotypeR renforcent la surveillance de routine de rhumes apparemment ordinaires qui, chez de nombreuses personnes, peuvent déclencher des maladies graves.
Citation: Luka, M.M., Nanjala, R., Rashed, W.M. et al. rhinotypeR enables reproducible rhinovirus genotype assignment from VP4/2 sequences. Sci Rep 16, 6149 (2026). https://doi.org/10.1038/s41598-026-37050-8
Mots-clés: génotypage des rhinovirus, surveillance moléculaire, ségrégation VP4/2, outils bioinformatiques, virus respiratoires