Clear Sky Science · fr
Évaluation comparative des méthodes de profilage transcriptomique ciblé HTG et TempO-Seq
Pourquoi c’est important pour la prise en charge du cancer
Lorsque médecins et chercheurs étudient le cancer, ils se tournent souvent vers les « molécules messagères » de la cellule — l’ARN — pour savoir quels gènes sont actifs ou silencieux. Ces profils peuvent révéler le comportement d’une tumeur et indiquer quels traitements pourraient être les plus efficaces. Mais la plupart des échantillons hospitaliers sont conservés dans des blocs de paraffine après fixation au formol, ce qui altère l’ARN fragile. Cette étude pose une question pratique aux conséquences importantes pour la recherche sur le cancer : maintenant qu’un test d’ARN largement utilisé a disparu du marché, une nouvelle méthode peut‑elle prendre le relais et fournir des résultats tout aussi utiles à partir de ces échantillons courants préservés ?
Deux outils pour lire l’activité génique
Pendant des années, de nombreux laboratoires se sont appuyés sur une méthode appelée HTG EdgeSeq Human Transcriptome Panel (HTP) pour lire l’activité des gènes directement à partir de petites coupes de tissu fixé au formol et inclus en paraffine (FFPE). Cette approche permettait d’examiner presque tous les gènes humains sans extraire préalablement l’ARN, ce qui économisait du temps et préservait la matière précieuse. Cependant, la société derrière HTG EdgeSeq a fait faillite, laissant les chercheurs à la recherche d’une alternative. Une technologie plus récente, TempO-Seq (TOS), proposée par un autre fabricant, promet des capacités similaires : elle cible également de nombreux gènes simultanément, fonctionne sur l’ARN dégradé des échantillons FFPE et est conçue pour être sensible, reproductible et relativement abordable.
Mettre les méthodes à l’épreuve
L’équipe de recherche a comparé ces deux technologies en face à face dans un contexte très pratique. Ils ont analysé 21 échantillons conservés de cancer de l’endomètre, ainsi que trois matériaux de référence ARN standard, d’abord avec HTG HTP puis avec TempO-Seq. Les deux méthodes utilisaient des panneaux ciblés qui, ensemble, couvraient plus de 18 000 des mêmes gènes. Les scientifiques ont appliqué des contrôles de qualité stricts, s’assurant que chaque échantillon produisait suffisamment de lectures de séquençage et que les mesures étaient stables. Ils ont aussi utilisé des outils statistiques pour supprimer les « effets de lot » — des différences artificielles pouvant survenir simplement parce que les tests ont été effectués à des jours, sur des machines ou des plateformes différentes.
Ce qui concorde et ce qui diverge
Lorsque l’équipe a examiné l’expression des gènes individuels un par un, les deux méthodes n’étaient pas toujours d’accord. Les différences dans la conception des sondes, la préparation des échantillons et le comptage des lectures peuvent rendre les comparaisons sur un seul gène bruyantes. Toutefois, la situation a changé lorsqu’ils ont étudié des motifs plus larges combinant les informations de nombreux gènes à la fois. Les signatures multi‑géniques — comme celles utilisées pour regrouper les tumeurs en sous‑types moléculaires, estimer la quantité de cellules immunitaires présentes dans un échantillon ou évaluer la pureté tumorale — montraient une bien meilleure concordance entre TempO‑Seq et HTG. Dans la plupart des cas, les scores ou classifications étaient similaires, même après que les chercheurs eurent simulé l’utilisation d’un nombre de lectures de séquençage inférieur pour imiter différentes capacités de machines.
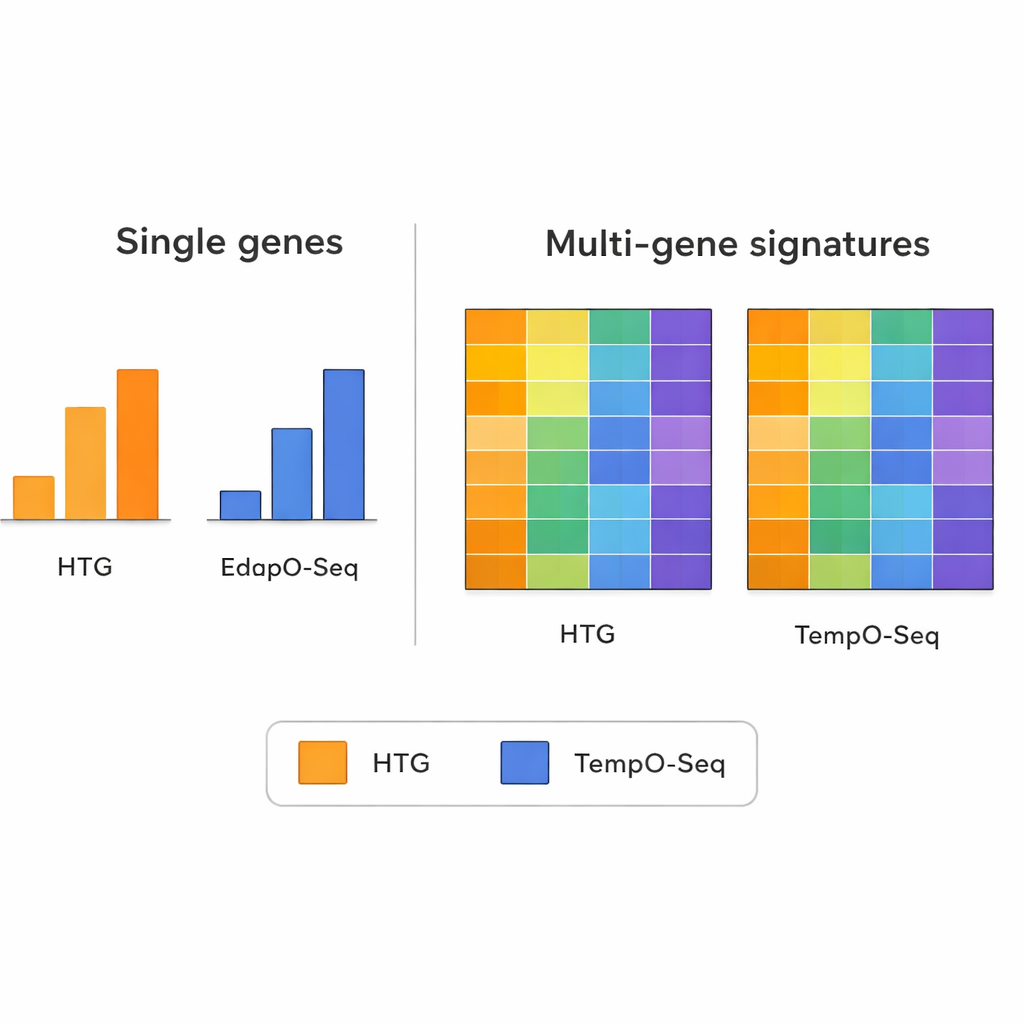
Les motifs multi‑géniques comme signaux fiables
L’étude met en évidence un principe important de la génomique moderne : si la mesure d’un gène isolé peut être perturbée par des artefacts techniques, la combinaison des signaux de dizaines ou de centaines de gènes tend à lisser ce bruit. Les auteurs ont utilisé plusieurs outils multi‑géniques bien connus comme tests de robustesse technique. Cela incluait un panel pour le cancer du sein qui classe les tumeurs en sous‑types intrinsèques, un algorithme évaluant la proportion de tissu immunitaire et conjonctif mêlée à un échantillon tumoral, et une méthode estimant les proportions de nombreux types cellulaires immunitaires. Sur ces lectures complexes, TempO‑Seq suivait généralement de près HTG, ce qui suggère qu’il capture les mêmes phénomènes biologiques même si certains détails fins diffèrent.
Ce que cela signifie pour l’avenir
Pour les chercheurs qui dépendent des archives FFPE pour étudier le cancer, la perte d’une plateforme de confiance aurait pu représenter un recul majeur. Cette étude de référence rassure : TempO‑Seq semble être un remplacement solide pour HTG HTP lorsque l’objectif est d’utiliser des biomarqueurs multi‑géniques et des profils d’expression larges, qui constituent l’épine dorsale de nombreux outils diagnostiques et pronostiques modernes. Les auteurs mettent en garde contre la comparaison directe des résultats d’un seul gène entre plateformes, car chaque méthode cible les gènes de manière légèrement différente. Ils recommandent plutôt de se concentrer sur des signatures complexes et multi‑géniques pour les études inter‑plateformes. En termes simples, la nouvelle méthode paraît capable de poursuivre le travail de sa prédécesseure pour la plupart des besoins réels de la recherche en oncologie, surtout lorsque les scientifiques s’intéressent au schéma global de nombreux gènes plutôt qu’à la valeur exacte d’un seul.
Citation: Fernández-Serra, A., López-Reig, R., Romero, I. et al. Comparative evaluation of HTG and TempO Seq targeted transcriptome profiling methods. Sci Rep 16, 6108 (2026). https://doi.org/10.1038/s41598-026-36810-w
Mots-clés: profilage transcriptomique, cancer de l’endomètre, tissu FFPE, séquençage ciblé de l’ARN, biomarqueurs d’expression génique