Clear Sky Science · fr
Une approche par apprentissage automatique pour prédire les coefficients osmotiques et déterminer les coefficients d'activité dans les sels d'ammonium alkyles
Des produits courants à la complexité cachée
Des assouplissants et après‑shampoings aux lingettes désinfectantes et bains de bouche, une famille de composés appelés sels d'ammonium quaternaires — souvent abrégés en « Quats » — est discrètement présente dans de nombreux produits que nous utilisons. Ils aident à éliminer les germes, assouplir les tissus et accélérer des réactions industrielles. Pourtant, prédire précisément le comportement de ces sels en solution aqueuse s'est avéré étonnamment difficile, ce qui limite la conception efficace de formulations plus sûres et plus écologiques. Cette étude montre comment l'apprentissage automatique moderne peut tirer parti des mesures passées pour prédire ce comportement de manière plus flexible et, dans de nombreux cas, plus précise que les modèles traditionnels.
Pourquoi ces sels comptent
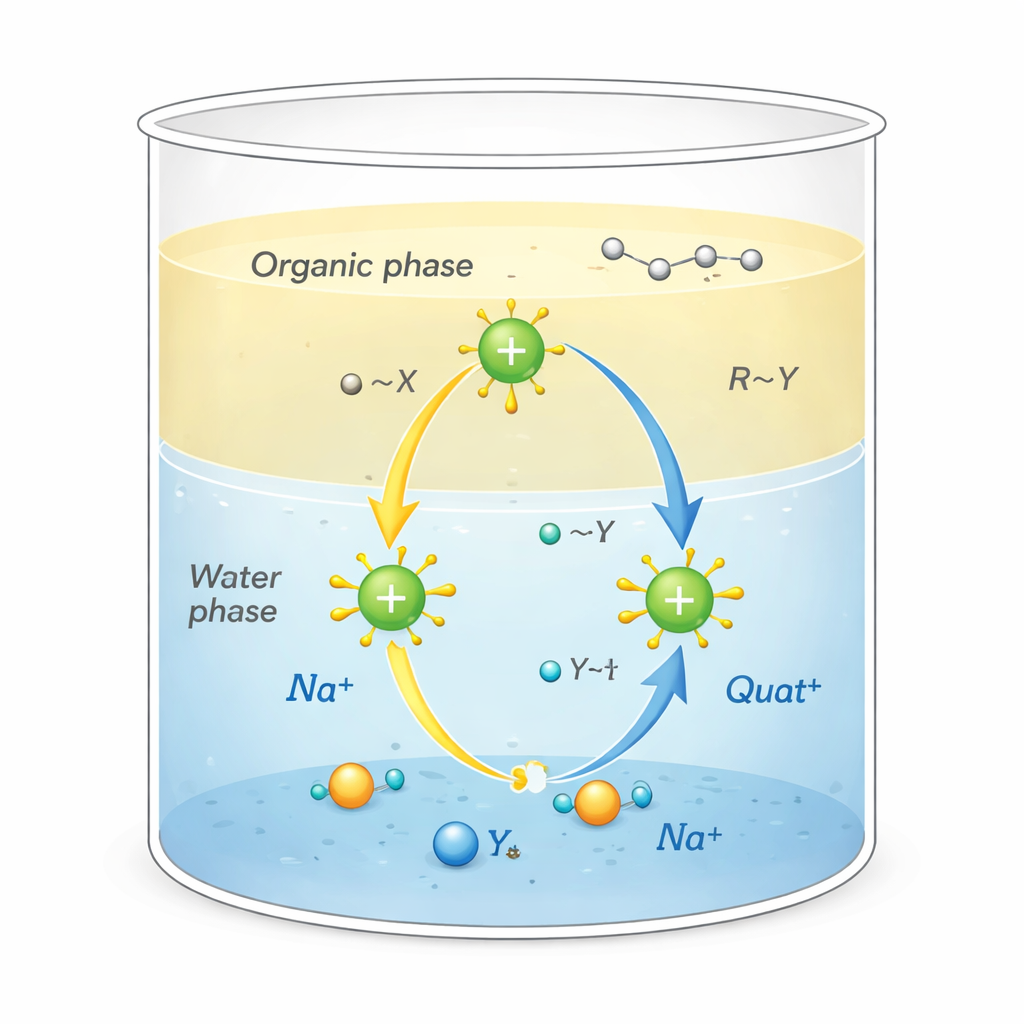
Les Quats sont des molécules chargées positivement entourées de « queues » riches en carbone. Cette forme particulière leur permet d'accomplir plusieurs fonctions à la fois : adhérer aux saletés grasses, rester accrochés à des surfaces comme les tissus ou les cheveux, et perturber les membranes des micro‑organismes, ce qui en fait de puissants désinfectants et tensioactifs. Ils sont également utilisés comme catalyseurs de transfert de phase, jouant le rôle de navettes qui transportent des ions réactifs de l'eau vers des solvants de type huileux où ces ions n'iraient pas naturellement. Ce transfert, qui se produit à l'interface eau/huile, peut accélérer de manière spectaculaire des réactions chimiques utilisées dans la fabrication de produits pharmaceutiques, de polymères et de produits fins.
Pourquoi il est difficile de prédire leur comportement
Pour concevoir de nouveaux Quats ou ajuster ceux qui existent, les chimistes doivent connaître leur comportement en solution — à quel point ils interagissent avec l'eau et avec d'autres ions dissous. Deux grandeurs clés sont le coefficient osmotique, qui reflète comment les sels influencent la tendance de l'eau à traverser des membranes, et le coefficient d'activité, qui mesure l'« efficacité » d'une espèce dissoute par rapport à une solution idéale parfaitement mélangée. Traditionnellement, ces valeurs sont obtenues soit par des expériences laborieuses, soit par des modèles physiques complexes tels que Electrolyte‑NRTL et Extended UNIQUAC, qui nécessitent de nombreux paramètres ajustés et sont difficiles à généraliser à de nouvelles molécules.
Apprendre à l'ordinateur à lire les molécules
Les chercheurs ont emprunté une voie différente : ils ont demandé si un ordinateur pouvait apprendre la relation entre la structure des Quats et leur comportement osmotique directement à partir de données existantes. Ils ont rassemblé 1 654 mesures de coefficients osmotiques pour 52 Quats différents dans la littérature scientifique. Chaque molécule a été décrite au moyen de la notation SMILES — une représentation textuelle qui encode des caractéristiques telles que le nombre d'atomes de carbone et d'oxygène, la présence d'anneaux benzéniques, le degré de ramification et le type de groupe d'azote chargé positivement, ainsi que l'anion associé (comme le chlorure, le bromure ou le nitrate). Ces descripteurs structuraux, ajoutés à la concentration du sel, ont servi d'entrées à plusieurs algorithmes d'apprentissage supervisé implémentés en Python.
Identifier le prédicteur le plus fiable
Sept algorithmes différents — régression linéaire, arbres de décision, forêts aléatoires, machines à vecteurs de support, gradient boosting, k‑plus proches voisins et processus gaussien — ont été entraînés sur 70 % des données et testés sur les 30 % restants. L'équipe a également utilisé un schéma de validation plus strict où toutes les données d'un même sel étaient exclues pour évaluer la capacité des modèles à extrapoler à un composé totalement inédit. La régression linéaire a donné de mauvais résultats, manquant des tendances non linéaires importantes. Les méthodes basées sur les arbres ont bien ajusté les données d'entraînement mais ont produit des prédictions légèrement irrégulières et ont perdu en précision sur de nouveaux sels. Le modèle par processus gaussien a trouvé le meilleur compromis : il a fourni des courbes lisses et physiquement plausibles pour les coefficients osmotiques et a obtenu une erreur absolue moyenne en pourcentage d'environ 5 % au global, dépassant les autres approches d'apprentissage automatique dans les tests les plus exigeants.
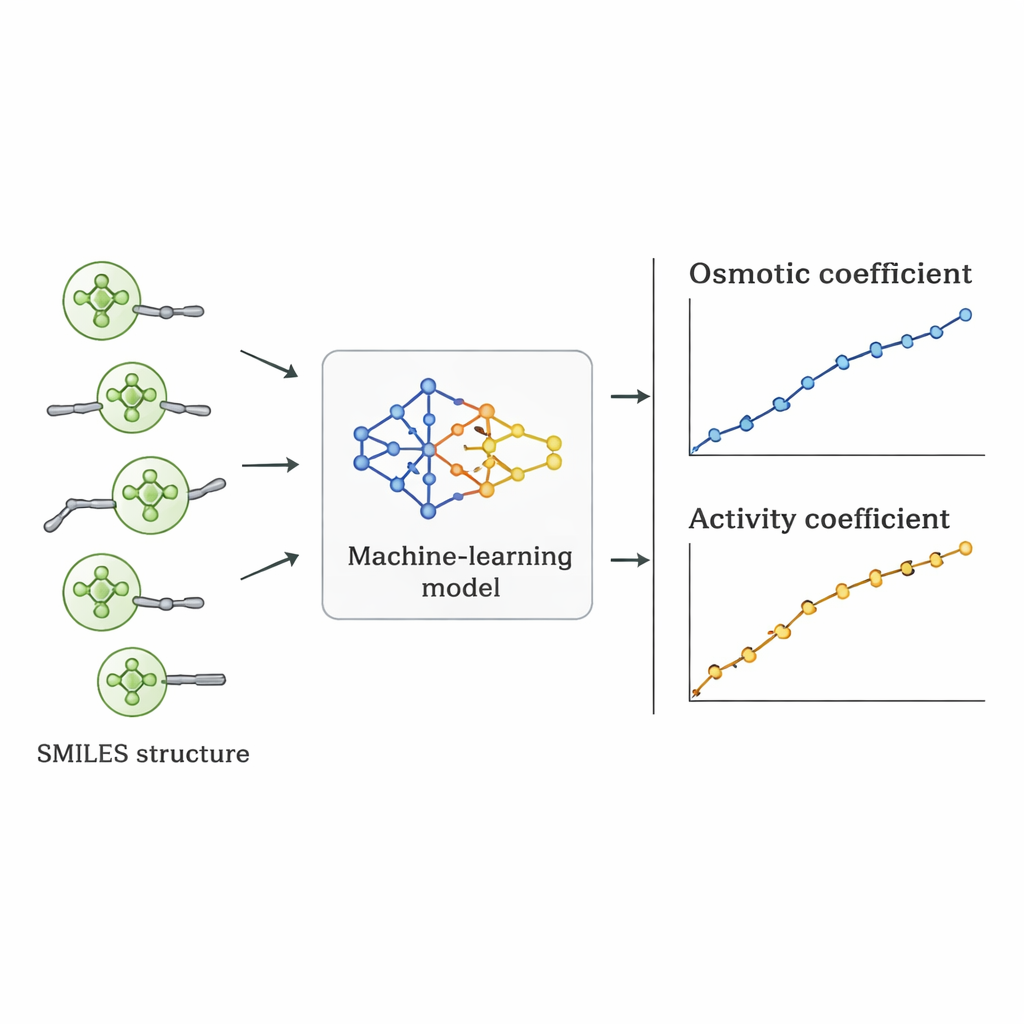
Du comportement osmotique à des nombres utiles pour la conception
Une fois le meilleur modèle sélectionné, ses coefficients osmotiques prédits ont été convertis en coefficients d'activité en utilisant des relations thermodynamiques standard. Lorsque ces coefficients d'activité ont été comparés aux valeurs issues d'expériences et de modèles physiques établis, l'approche par apprentissage automatique les a souvent égalées ou surpassées pour certains Quats. Bien que son erreur moyenne sur l'ensemble des substances fût légèrement supérieure à celle de certains modèles spécialisés, elle présente un avantage crucial : étant pilotée par des descripteurs structurels plutôt que par un ajustement propre à chaque sel, elle peut s'appliquer à de nouveaux Quats jamais mesurés en laboratoire, pourvu que leurs structures ressemblent à celles présentes dans l'ensemble d'entraînement.
Quelles implications pour les produits et les procédés
Pour un non‑spécialiste, le message est que les ordinateurs peuvent désormais « lire » des descriptions textuelles compactes de molécules et, à partir de motifs appris dans des données antérieures, prédire avec une précision remarquable comment ces molécules se comporteront en milieu aqueux. Cela ouvre la voie à un criblage plus rapide et moins coûteux de nouveaux Quats pour les désinfectants, les nettoyants, les produits de soin personnel et les catalyseurs industriels, sans nécessiter d'expérimentations exhaustives pour chaque candidat. Le modèle actuel n'est qu'une première étape, et les auteurs notent que des empreintes moléculaires plus riches et des algorithmes plus récents pourraient encore améliorer les performances. Néanmoins, il illustre comment des outils fondés sur les données peuvent compléter la chimie traditionnelle, aidant les ingénieurs à concevoir des formulations plus efficaces et potentiellement plus sûres en explorant des possibilités chimiques qu'il serait impraticable de tester une à une en laboratoire.
Citation: Chawuthai, R., Murathathunyaluk, S., Saengsuradech, S. et al. A machine learning approach for predicting osmotic coefficients and deriving activity coefficients in alkyl ammonium salts. Sci Rep 16, 5969 (2026). https://doi.org/10.1038/s41598-026-36758-x
Mots-clés: sels d'ammonium quaternaires, catalyse par transfert de phase, coefficients osmotiques, coefficients d'activité, apprentissage automatique en chimie