Clear Sky Science · fr
Cadre hybride d'apprentissage profond pour une classification précise de données génomiques à haute dimension
Donner du sens à la déferlante de données génomiques
Les technologies modernes d’analyse de l’ADN peuvent mesurer des dizaines de milliers de gènes en une seule expérience, ouvrant la voie à une détection plus précoce des maladies et à des traitements plus précis. Pourtant, cette abondance de données est si volumineuse, bruyante et complexe que même des modèles informatiques puissants peinent souvent à identifier des motifs clairs et fiables. Cet article présente un nouveau type de système d’intelligence artificielle (IA) conçu spécifiquement pour traiter des données génomiques écrasantes, visant à rendre les prédictions plus précises tout en expliquant comment ces prédictions ont été obtenues.
Pourquoi les données génomiques sont si difficiles à exploiter
Les études génomiques produisent routinièrement beaucoup plus de mesures qu’il n’y a de patients ou d’échantillons. Nombre de ces mesures sont non pertinentes, redondantes ou déformées par le bruit technique. Les méthodes classiques d’apprentissage automatique exigent soit que des experts humains sélectionnent manuellement les gènes potentiellement importants, soit qu’on utilise tout et qu’on prenne le risque de surapprentissage — c’est‑à‑dire de bien fonctionner sur les données d’entraînement mais d’échouer sur de nouveaux cas. L’apprentissage profond, qui a transformé des domaines comme la reconnaissance d’images, peut apprendre automatiquement des motifs à partir de données brutes. Cependant, en génomique, il se comporte souvent comme une boîte noire : il peut fournir des réponses précises, mais livre peu d’éléments expliquant le pourquoi, ce qui limite son adoption en médecine où la transparence est essentielle.
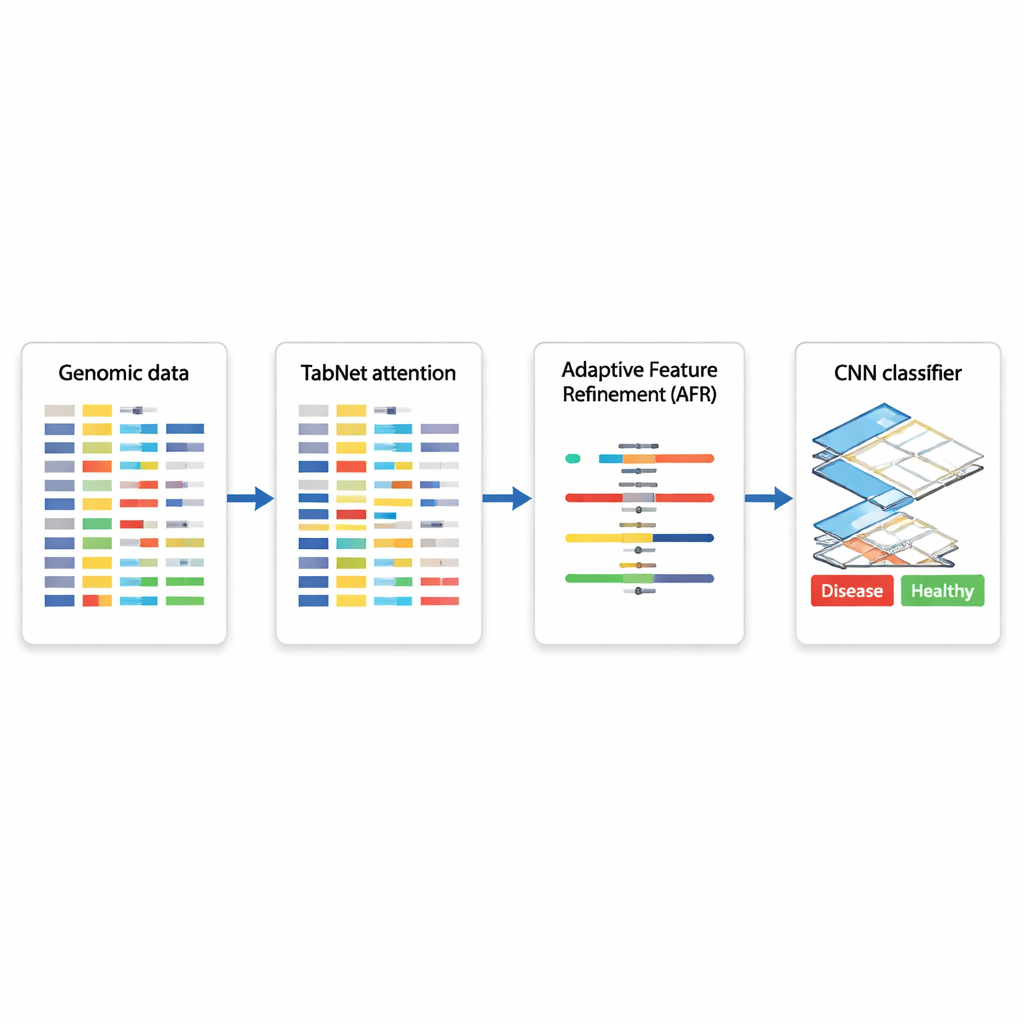
Une architecture IA hybride pour les décisions basées sur les gènes
Les auteurs proposent une architecture d’apprentissage profond hybride qui enchaîne trois modules spécialisés. D’abord, un composant appelé TabNet joue le rôle d’un projecteur, parcourant toutes les mesures génomiques disponibles et apprenant quelles caractéristiques sont les plus informatives pour une tâche donnée — par exemple, distinguer un tissu cancéreux d’un tissu non cancéreux. Plutôt que de traiter chaque gène de manière égale, TabNet concentre l’attention sur un sous‑ensemble parcimonieux jugé le plus pertinent. Ensuite, une couche de Raffinement Adaptatif des Caractéristiques (AFR) prend ces signaux sélectionnés et les repondère, renforçant les motifs cohérents et significatifs tout en atténuant davantage le bruit. Enfin, un réseau de neurones convolutionnel (CNN), couramment utilisé en analyse d’images, examine comment les caractéristiques raffinées interagissent localement, capturant les relations subtiles entre groupes de gènes susceptibles d’indiquer un sous‑type de maladie ou un état biologique particulier.
Évaluation du modèle
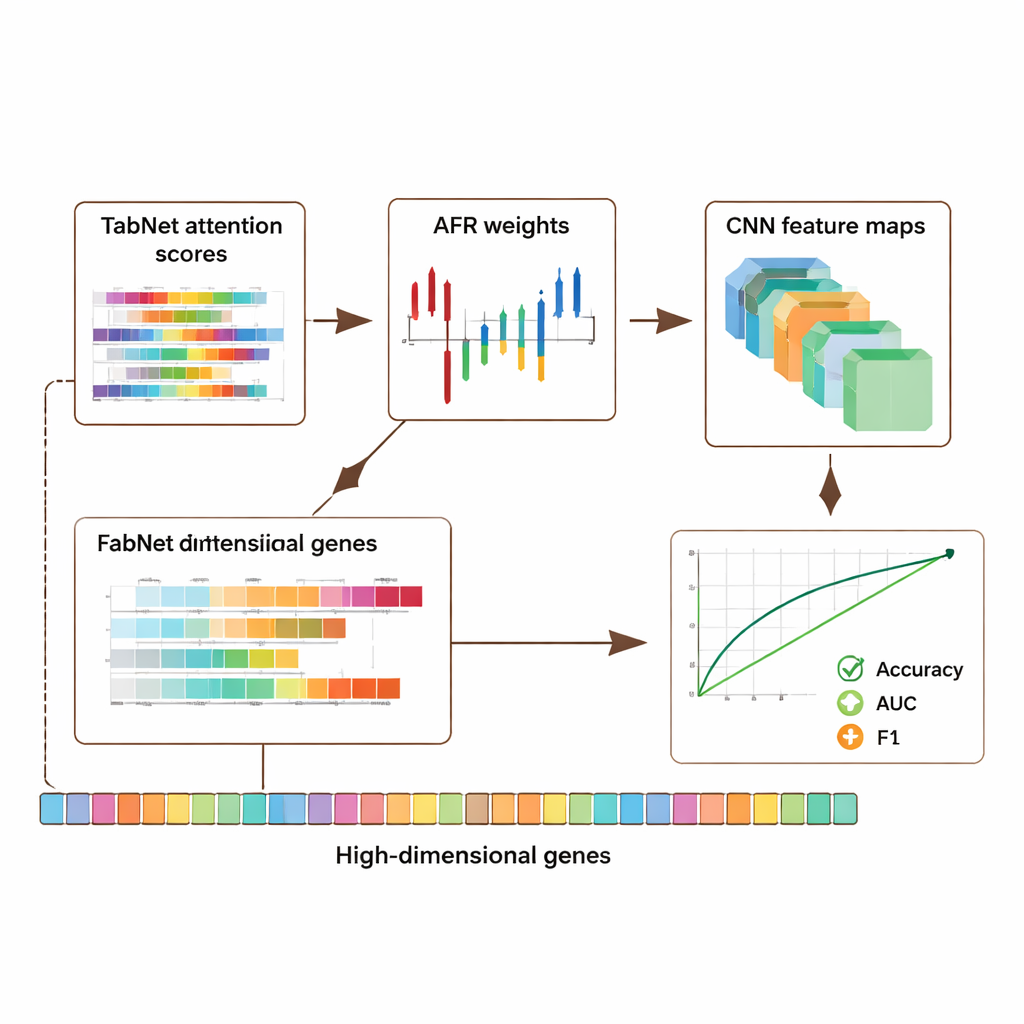
Le cadre a été évalué sur trois ressources publiques majeures : un jeu de données sur le cancer du sein provenant du Cancer Genome Atlas, un jeu de données de mélanome en cellule unique issu du Gene Expression Omnibus, et un jeu de données épigénomiques du projet ENCODE. Ensemble, ces collections comprennent des milliers d’échantillons et des dizaines de milliers de caractéristiques par échantillon, couvrant l’activité génique et les marques chimiques sur l’ADN. Sur l’ensemble des jeux de données, le modèle hybride a surpassé plusieurs approches de pointe, améliorant la précision et des mesures clés de qualité de classification telles que l’aire sous la courbe ROC (AUC) et le F1‑score d’environ 5 à 8 points de pourcentage. Fait important, ces gains ne se sont pas faits au détriment de la transparence : le modèle produit des cartes d’attention via TabNet et des cartes d’activation via le CNN qui mettent en évidence les gènes et régions les plus influents dans chaque prédiction.
Équilibrer précision, confidentialité et confiance
Parce que les données génomiques sont profondément personnelles, les auteurs ont également étudié comment protéger la confidentialité tout en conservant un signal utile. Ils ont introduit un mécanisme de confidentialité adaptatif qui ajoute plus de bruit aux caractéristiques très sensibles et moins aux autres, combiné à un masquage de certaines entrées sélectionnées. Les tests ont montré que même lorsque un bruit modéré était introduit, le modèle conservait une forte précision et capacité de discrimination, la performance se dégradant de façon progressive au fur et à mesure que la protection était renforcée. Parallèlement, les schémas d’attention et d’activation interprétables pointaient souvent vers des gènes déjà connus pour jouer un rôle dans le cancer et la régulation immunitaire, suggérant que le système ne se contente pas de mémoriser les données mais capture des signaux biologiquement pertinents. Une étude d’ablation — suppression systématique de parties de l’architecture — a confirmé que chaque module, en particulier la couche AFR, apportait une contribution mesurable à la performance.
Ce que cela signifie pour la médecine de demain
Concrètement, ce travail propose une manière plus intelligente de filtrer d’énormes tableaux génomiques pour trouver des motifs liés à la maladie, tout en indiquant quelles lignes du tableau ont le plus compté. En combinant sélection ciblée des caractéristiques, raffinement soigné et reconnaissance de motifs, le modèle hybride améliore la précision des prédictions, reste gérable en termes de calcul et fournit des indices visuels interprétables par cliniciens et biologistes. Bien que des tests supplémentaires soient nécessaires sur des populations de patients plus larges et diversifiées, de tels cadres pourraient aider à identifier de nouveaux biomarqueurs, affiner des sous‑types de maladies et soutenir des outils d’aide à la décision clinique en médecine de précision — rapprochant l’analyse IA de l’ADN de l’usage réel en clinique.
Citation: Swain, M.K., Kamila, N.K., Jena, L. et al. Hybrid deep learning framework for accurate classification of high dimensional genomic data. Sci Rep 16, 5919 (2026). https://doi.org/10.1038/s41598-026-36128-7
Mots-clés: apprentissage profond génomique, découverte de biomarqueurs du cancer, IA interprétable, médecine de précision, génomique préservant la vie privée