Clear Sky Science · fr
Une étude quantitative des composés cytotoxiques utilisant des descripteurs basés sur des graphes et l’apprentissage automatique
Pourquoi cette recherche compte pour les futurs médicaments anticancéreux
Les médicaments anticancéreux qui tuent les cellules tumorales, appelés médicaments cytotoxiques, naviguent souvent sur une ligne fragile entre sauver des vies et provoquer des effets secondaires graves. Pour concevoir des traitements plus sûrs et plus efficaces, les chercheurs ont besoin de moyens rapides et fiables pour prédire comment ces molécules se déplacent dans l’organisme — à quel point elles sont absorbées, leur capacité à traverser les membranes cellulaires et où elles se distribuent. Cette étude montre comment des descriptions mathématiques des molécules, combinées à l’apprentissage automatique moderne, peuvent estimer avec précision une propriété clé qui régule ce comportement, ce qui pourrait accélérer la recherche de meilleures thérapies anticancéreuses.
Une surface clé qui contrôle où les médicaments peuvent aller
Une idée centrale de l’article est la surface polaire topologique, ou Top_PSA. En termes simples, il s’agit d’un nombre qui reflète quelle proportion de la surface d’une molécule est constituée de régions « polaires » — des parties qui apprécient l’eau et peuvent former des liaisons hydrogène. Les molécules présentant des surfaces polaires très élevées ont souvent du mal à traverser les membranes lipidiques et peuvent être mal absorbées par voie orale. À l’inverse, les molécules avec des surfaces polaires très faibles peuvent franchir trop facilement de nombreuses barrières, provoquant parfois des effets indésirables dans des tissus sensibles comme le cerveau. La Top_PSA est devenue un raccourci populaire pour estimer ces propriétés de transport parce qu’elle peut être calculée rapidement à partir d’un dessin 2D de la molécule, sans nécessiter de simulations 3D lentes.
Transformer des dessins moléculaires en nombres
Les chercheurs ont constitué un jeu de données soigné de 156 composés cytotoxiques différents, issus de médicaments anticancéreux réels et d’agents expérimentaux. Ils ont ensuite converti chaque molécule en 58 descripteurs dits — des nombres qui capturent des caractéristiques telles que le nombre d’atomes, le nombre d’anneaux, la flexibilité des liaisons, le nombre d’atomes capables de former des liaisons hydrogène, et le degré de polarité ou d’électronégativité de différentes parties. Beaucoup de ces descripteurs proviennent de la théorie des graphes, où une molécule est traitée comme un réseau de nœuds et de liens connectés. Ce portrait numérique riche de chaque molécule a servi d’entrée pour des modèles informatiques visant à prédire les valeurs de Top_PSA calculées par des boîtes à outils chimiques largement utilisées.
Tester plusieurs voies vers une prédiction précise
Pour trouver la meilleure façon de relier ces descripteurs à la Top_PSA, l’équipe a comparé plusieurs stratégies de modélisation. Ils ont testé la régression linéaire standard ainsi que deux variantes « régularisées » appelées ridge et LASSO, conçues pour mieux gérer l’information bruyante et redondante. Ils ont également exploré différents schémas de préparation des données : ajuster les modèles directement sur les descripteurs bruts, les compresser avec l’analyse en composantes principales (PCA), les mettre à l’échelle de manière à réduire l’impact des valeurs extrêmes (robust scaling), corriger les valeurs aberrantes et élaguer les caractéristiques fortement corrélées en utilisant une mesure appelée facteur d’inflation de la variance. Chaque approche a été évaluée soigneusement via une validation croisée en k plis, une méthode qui divise à répétition les données en sous-ensembles d’entraînement et de test pour se prémunir contre le surapprentissage.
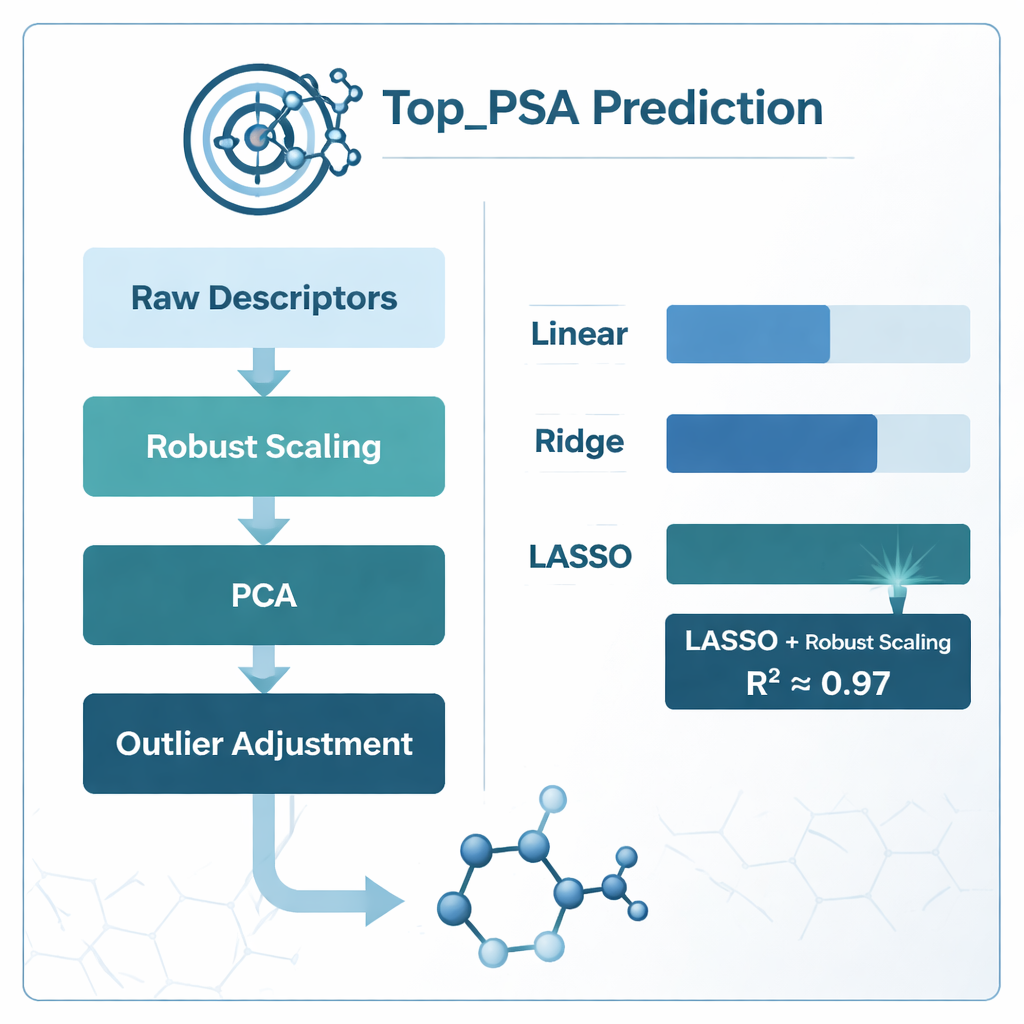
Ce qui a le mieux fonctionné et ce que les modèles ont appris
Le gagnant clair fut la combinaison de la mise à l’échelle robuste avec la régression LASSO, qui a atteint un coefficient de détermination (R²) d’environ 0,97 — ce qui signifie qu’elle expliquait approximativement 97 % de la variation de la Top_PSA sur les 156 molécules. Les modèles basés sur la PCA se sont approchés en précision brute mais étaient plus difficiles à interpréter chimiquement, car les descripteurs originaux y sont fusionnés en composantes abstraites. Une simple élagage des descripteurs corrélés à l’aide du facteur d’inflation de la variance a en fait dégradé les performances, ce qui suggère que certaines mesures redondantes contiennent encore des informations chimiques utiles. En examinant les poids des descripteurs que LASSO a conservés non nuls, les auteurs ont trouvé que les facteurs les plus importants étaient la présence d’hétéroatome tels que l’azote et l’oxygène, la capacité à donner ou accepter des liaisons hydrogène, et des indices qui suivent la disposition des atomes électronégatifs dans le graphe moléculaire — autant de caractéristiques cohérentes avec la compréhension chimique intuitive de la surface polaire.
Comment cela peut guider une meilleure conception des médicaments
Pour les lecteurs extérieurs au domaine, le message clé est que des empreintes mathématiques de molécules soigneusement préparées, lorsqu’elles sont associées à des méthodes d’apprentissage automatique bien choisies, peuvent fournir des estimations rapides et fiables de la propension des médicaments anticancéreux à « coller » ou « traverser » les barrières biologiques au cours de leur parcours dans l’organisme. L’étude offre des conseils pratiques aux autres chercheurs sur la manière de prétraiter les données de descripteurs, quelles approches de modélisation privilégier et quels raccourcis éviter. À long terme, de tels modèles robustes et interprétables de la Top_PSA peuvent aider les chimistes à filtrer d’immenses bibliothèques virtuelles de candidats-médicaments, en concentrant les efforts sur les composés présentant le bon équilibre entre perméation membranaire et sécurité — une étape importante vers des traitements anticancéreux plus efficaces et moins toxiques.
Citation: Ahmad, S., Javed, S., Khalid, S. et al. A quantitative study of cytotoxic compounds using graph based descriptors and machine learning. Sci Rep 16, 5076 (2026). https://doi.org/10.1038/s41598-026-35728-7
Mots-clés: médicaments cytotoxiques, surface polaire, descripteurs moléculaires, apprentissage automatique, perméabilité des médicaments