Clear Sky Science · fr
Dysfonction mitochondriale et dérégulation du Ca2+ dans des neurones humains dérivés d’iPSC porteurs d’une mutation de la préséniline‑1 apparaissent sous stress via un mécanisme indépendant de MCU‑1
Pourquoi cela importe pour la maladie d’Alzheimer
La maladie d’Alzheimer est souvent décrite en termes de plaques protéiques collantes dans le cerveau, mais bien avant que la mémoire ne décline, les minuscules « centrales » à l’intérieur des neurones — les mitochondries — et la gestion des ions calcium peuvent déjà dysfonctionner. Cette étude utilise des neurones humains cultivés à partir de cellules cutanées d’une personne porteuse d’une mutation familiale bien connue pour poser une question simple mais cruciale : à quel moment et de quelle manière la production d’énergie et l’homéostasie calcique commencent‑elles à faillir ?
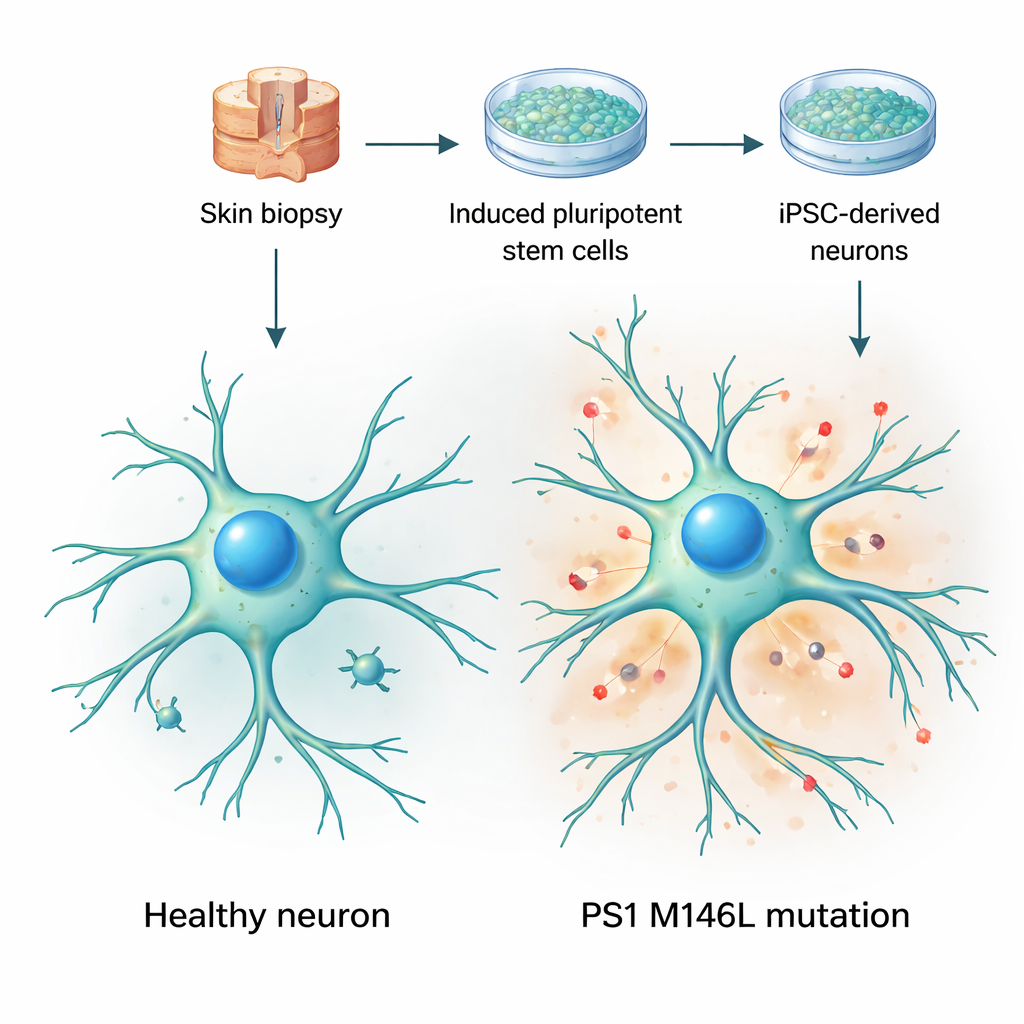
Transformer des cellules de peau en modèles cérébraux vivants
Les chercheurs ont commencé par des biopsies de peau prélevées chez deux femmes : une volontaire en bonne santé et une porteuse sans symptômes d’une mutation de la préséniline‑1 nommée M146L, présente dans une famille argentine atteinte d’un Alzheimer à début précoce. Ils ont reprogrammés les cellules cutanées en cellules souches pluripotentes induites — des cellules capables de devenir presque n’importe quel tissu — puis les ont orientées vers un destin neuronal. Au cours de plusieurs semaines en culture, ces cellules ont acquis des formes neuronales typiques, étendu de longs prolongements ramifiés et exprimé des marqueurs neuronaux standards. Fait important, les cellules contrôle et mutantes ont mûri à des rythmes similaires et présentaient un aspect globalement sain, ce qui a permis à l’équipe de se concentrer sur des changements fonctionnels subtils plutôt que sur une perte cellulaire évidente ou des dommages apparents.
Signaux électriques et calcium sous tension
Les neurones dépendent d’un contrôle strict du calcium, un atome chargé qui agit comme un interrupteur rapide pour de nombreux processus cellulaires. À l’aide de sondes fluorescentes, l’équipe a suivi les variations de calcium intracellulaire lors de stimulation électrique par le potassium ou d’activation par des molécules de signalisation. Lors d’une simple dépolarisation, les neurones porteurs de la mutation M146L montraient des augmentations de calcium plus faibles que les neurones témoins, suggérant des difficultés à maintenir les gradients électriques et ioniques qui favorisent normalement l’entrée de calcium. Cependant, lorsque les chercheurs ont provoqué une situation plus stressante — forçant la fuite de calcium depuis les réserves internes du réticulum endoplasmique — la différence est devenue plus marquée. En réponse à ce stress, les mitochondries des neurones mutants ont capté sensiblement moins de calcium que celles des cellules témoins, indiquant une capacité réduite à tamponner des pics calciques potentiellement dangereux.
Dissocier l’utilisation d’énergie de l’équilibre calcique
Pour comprendre comment cette gestion altérée du calcium affecte le métabolisme cellulaire, les investigateurs ont mesuré la consommation d’oxygène des neurones — un proxy direct de l’activité mitochondriale. Fait surprenant, les neurones portant la mutation M146L consommaient davantage d’oxygène : leurs taux de consommation basale et maximale d’oxygène, ainsi que la part d’oxygène liée à la production d’ATP, étaient tous plus élevés que chez les témoins. Pourtant, l’efficacité de couplage entre consommation d’oxygène et production d’ATP semblait similaire, et il n’y avait pas d’augmentation du nombre de mitochondries ni des enzymes clés de fabrication d’ATP. En revanche, les mitochondries des neurones mutants étaient plus longues et plus tubulaires, avec des niveaux plus élevés d’une protéine de fusion appelée mitofusine‑1, un profil souvent observé dans des cellules soumises à un stress chronique de faible intensité. Ces mitochondries hyperactives et allongées généraient également davantage d’espèces réactives de l’oxygène, des molécules instables pouvant endommager protéines et ADN si elles ne sont pas contrôlées.
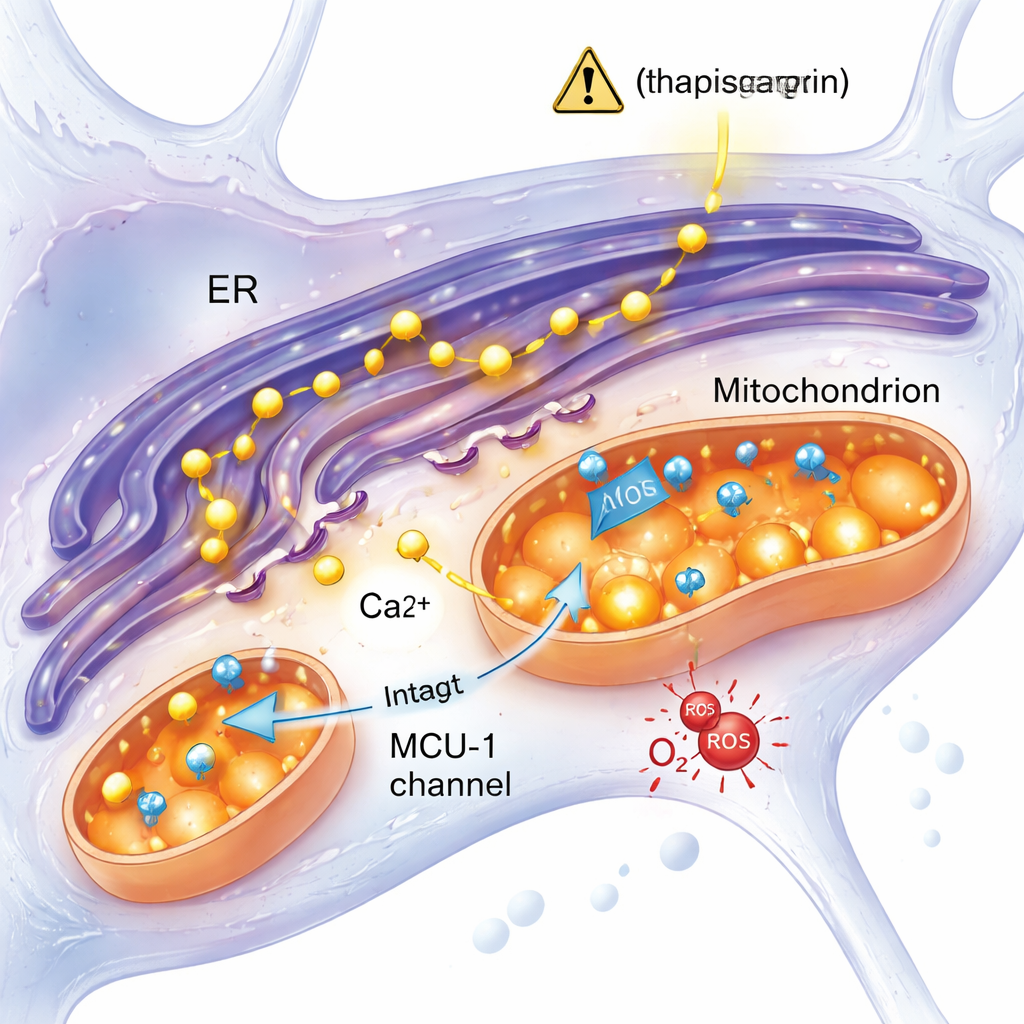
Une réponse au stress indépendante d’un canal calcique clé
Une hypothèse majeure en recherche sur Alzheimer est que l’excès de calcium provenant du réticulum endoplasmique se précipite dans les mitochondries via un canal appelé uniporteur calcique mitochondrial (MCU‑1), les surchargeant et provoquant leur dysfonction. Cette étude a testé directement cette idée. Lorsque l’équipe a bloqué MCU‑1 avec un inhibiteur spécifique, les neurones contrôle et mutants ont montré de fortes réductions de l’entrée calcique mitochondriale, confirmant que le canal fonctionnait dans les deux groupes. De plus, lorsque la libération de calcium a été déclenchée via une voie plus physiologique impliquant le récepteur IP3 — une autre porte calcique clé — les cellules mutantes et témoins ont répondu de façon similaire. Ces résultats écartent l’hypothèse d’un MCU‑1 défectueux et suggèrent plutôt que les contacts physiques et fonctionnels entre réticulum endoplasmique et mitochondries, ou d’autres aspects de leur interaction, sont altérés dans les neurones mutants.
Ce que cela signifie pour la compréhension et le traitement de la maladie
Pris ensemble, les résultats dressent le portrait de neurones humains porteurs de la mutation PS1 M146L qui semblent normaux au repos mais réagissent anormalement sous stress. Leurs mitochondries n’absorbent pas suffisamment de calcium lorsque les réserves internes sont soudainement libérées, pourtant elles tournent plus intensément — consommant plus d’oxygène et générant plus d’espèces réactives — comme si elles étaient verrouillées dans un mode compensatoire coûteux. Parce que cela se produit dans des neurones humains dérivés, avant tout symptôme clinique, le travail soutient l’idée que la perturbation de la signalisation calcique et la suractivité mitochondriale précoce sont des événements en amont dans la maladie d’Alzheimer, et non de simples sous‑produits tardifs. Pour le grand public, le message clé est que maintenir l’équilibre entre signaux calciques et production d’énergie mitochondriale peut être aussi central pour prévenir la maladie que de cibler les plaques amyloïdes mieux connues.
Citation: Wilson, C., Galeano, P., Remedi, M.M. et al. Mitochondrial dysfunction and Ca2+ dysregulation in human iPSC-derived neurons carrying presenilin-1 mutation arise under stress via an MCU-1-independent mechanism. Sci Rep 16, 6002 (2026). https://doi.org/10.1038/s41598-026-35597-0
Mots-clés: Maladie d’Alzheimer, mitochondries, signalisation calcique, mutation de la préséniline‑1, neurones dérivés d’iPSC