Clear Sky Science · fr
Développement structuro‑computationnel d’analogues de 2,5‑disubstitués‑1,3,4‑oxadiazole en tant qu’inhibiteurs actifs de LOX, d’uréase et d’α‑glucosidase
Nouveaux outils chimiques pour lutter contre des maladies courantes
De nombreux problèmes de santé quotidiens — des ulcères et calculs rénaux au diabète et à l’inflammation chronique — sont alimentés par une activité excessive d’enzymes dans l’organisme. Cette étude explore une famille de petites molécules récemment conçues qui agissent comme de minuscules freins pour trois de ces enzymes. En ajustant leurs structures et en les testant en laboratoire et par modélisation informatique, les chercheurs visent à poser les bases de médicaments futurs plus efficaces et plus sûrs. 
Pourquoi ces enzymes sont importantes
L’équipe s’est concentrée sur trois cibles enzymatiques qui jouent des rôles très différents mais tout aussi cruciaux pour la santé. L’uréase aide à décomposer l’urée ; lorsqu’elle devient hyperactive chez certaines bactéries ou dans certains tissus, elle peut contribuer aux ulcères gastriques et urinaires, aux calculs rénaux et même à l’hypertension. L’α‑glucosidase se trouve à la surface des cellules intestinales et coupe les glucides complexes en glucose ; la bloquer est une stratégie bien établie pour aider à contrôler la glycémie dans le diabète de type 2. La lipoxygénase (LOX) convertit les lipides en molécules de signalisation qui favorisent l’inflammation, l’asthme et certains processus liés au cancer. On recherche donc des médicaments capables de réduire l’activité de ces enzymes sans nuire à d’autres systèmes.
Constitution d’une bibliothèque de molécules candidates
Pour chercher de tels agents, les chercheurs ont synthétisé une série de 15 composés apparentés basés sur un système cyclique appelé 1,3,4‑oxadiazole relié à une unité pipéridine. Cet échafaudage chimique figure déjà dans plusieurs médicaments modernes et interagit bien avec des cibles biologiques. L’équipe a modifié les « ornements » attachés à ce cœur — petits groupes tels que méthyle, éthyle, méthoxy, benzyle et cyclohexyle — à diverses positions, créant une petite bibliothèque de candidats étiquetés 7a à 7o. Ils ont confirmé chaque structure à l’aide d’outils analytiques standards comme la spectroscopie infrarouge et la résonance magnétique nucléaire, s’assurant que les produits obtenus correspondaient aux conceptions.
Évaluation de la capacité des molécules à inhiber les enzymes
Chaque composé a ensuite été testé sur la LOX purifiée, l’uréase et l’α‑glucosidase afin d’évaluer sa capacité à ralentir l’activité enzymatique. Plusieurs molécules se sont distinguées. Contre l’α‑glucosidase, les composés 7a et 7n se sont montrés particulièrement efficaces, surpassant le médicament antidiabétique de référence acarbose dans les tests in vitro en atteignant des pourcentages d’inhibition élevés à de faibles concentrations micromolaires. Pour la LOX, les composés 7a, 7h et 7n ont affiché une activité très forte, parfois meilleure que l’inhibiteur naturel de référence quercétine. Dans les essais sur l’uréase, les composés 7a et surtout 7l se sont montrés comparables voire légèrement supérieurs à l’inhibiteur standard thiourée, suggérant qu’ils pourraient à terme servir de pistes pour des thérapies anti‑ulcère ou anti‑calculs. 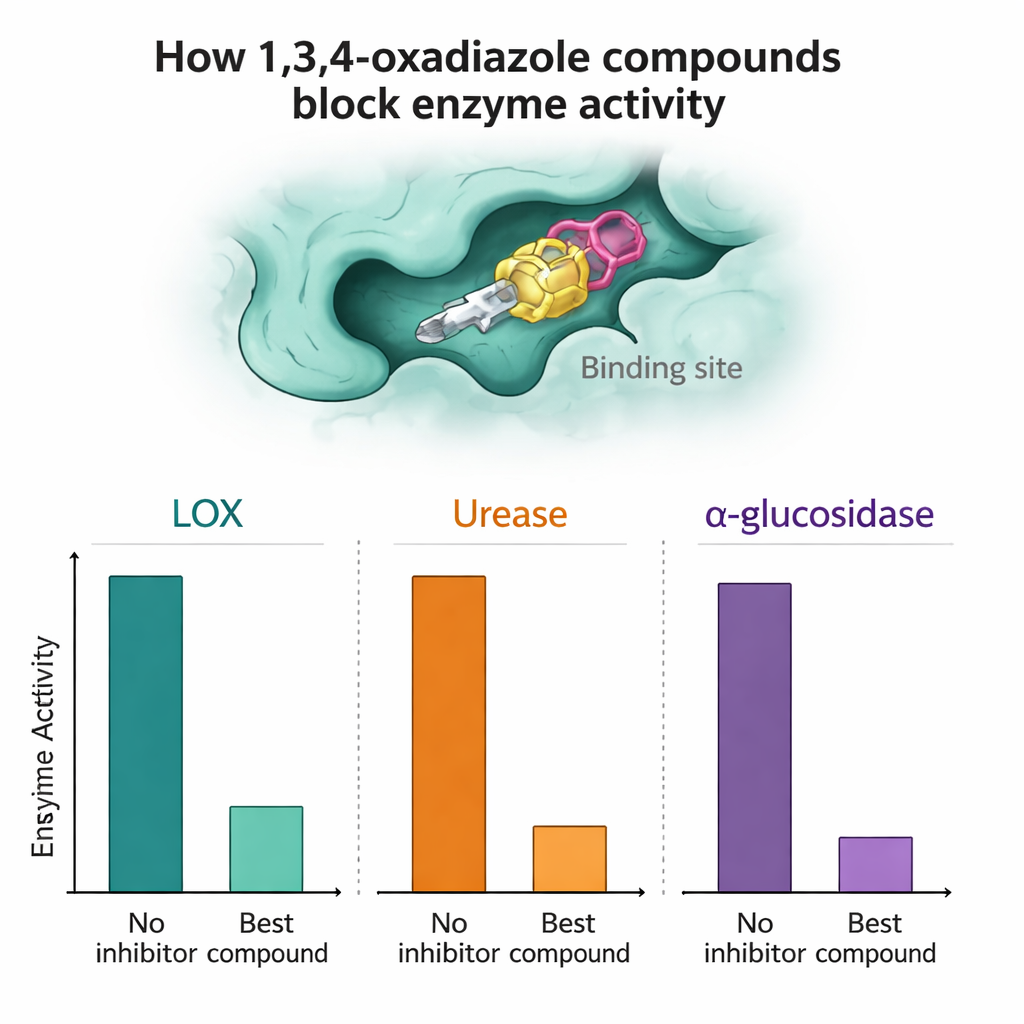
Relier la structure à la performance
Puisque les différences entre les composés 7a–7o se limitent à de petites modifications autour du noyau, les chercheurs ont pu commencer à cartographier de simples relations structure‑activité. Par exemple, l’ajout de petits groupes riches en carbone à certaines positions « ortho » d’un anneau attaché favorisait souvent la capacité à bloquer l’α‑glucosidase ou la LOX. D’autres substitutions, comme certains groupes méthoxy ou esters placés moins favorablement, avaient tendance à affaiblir l’activité. Pour comprendre pourquoi, l’équipe a eu recours à des simulations informatiques. Des calculs quantiques ont montré que toutes les molécules étaient thermodynamiquement stables et suffisamment flexibles pour s’adapter aux cavités enzymatiques. Des études de docking — ajustement virtuel de chaque molécule dans des modèles tridimensionnels des enzymes — ont révélé que les composés les plus actifs établissaient des contacts plus étroits via un mélange de liaisons hydrogène et d’interactions hydrophobes à des positions clés des sites actifs, alors que les candidats moins puissants se logeaient moins bien ou manquaient des points de contact critiques.
Ce que cela signifie pour les médicaments de demain
En termes concrets, l’étude identifie une poignée de « clés » prometteuses qui s’ajustent bien à trois « serrures » liées à la maladie et commence à expliquer, au niveau atomique, pourquoi certaines clés fonctionnent mieux que d’autres. Aucune de ces molécules n’est encore prête à devenir un médicament — elles doivent encore faire l’objet de tests de sécurité, d’optimisations pour leur comportement dans l’organisme et d’essais chez l’animal et l’humain. Mais le travail montre que le cadre 1,3,4‑oxadiazole–pipéridine constitue un point de départ fertile pour des traitements ciblant le diabète, les affections inflammatoires et les troubles liés à l’uréase. La combinaison de mesures expérimentales soignées et de modélisation informatique détaillée offre une feuille de route pour affiner ces candidats en médicaments plus précis et plus efficaces.
Citation: Javid, J., Aziz-ur-Rehman, Iqbal, J. et al. Structural and computational supported development of 2,5-disubstituted-1,3,4-oxadiazole analogues as active LOX, urease, and α-glucosidase inhibitors. Sci Rep 16, 5866 (2026). https://doi.org/10.1038/s41598-026-35499-1
Mots-clés: inhibiteurs d’enzymes, découverte de médicaments, composés oxadiazoles, diabète et inflammation, urée et lipoxygénase