Clear Sky Science · fr
Perturbation de l’homéostasie du fer intracellulaire par dysfonction mitochondriale associée à la suppression de l’expression d’ATP13A2
Pourquoi le fer à l’intérieur des cellules cérébrales compte
La maladie de Parkinson est surtout connue pour ses tremblements et la rigidité des mouvements, mais au cœur des neurones affectés se joue un autre drame : le fer, métal essentiel, commence à s’accumuler là où il ne devrait pas. Cette étude pose une question simple mais importante : comment cet excès de fer se produit‑il, et comment peut‑il endommager les petites centrales énergétiques et les centres de recyclage des cellules nerveuses ? En y répondant, le travail donne des pistes sur les raisons de la dégénérescence de certaines régions cérébrales dans la maladie de Parkinson et les troubles apparentés, et ouvre la voie à de nouveaux types de traitements qui vont au‑delà du simple remplacement de la dopamine.
Un examen approfondi d’un indice génétique rare
Les chercheurs se concentrent sur une forme héréditaire rare de la maladie de Parkinson, appelée PARK9, due à des anomalies d’un gène nommé ATP13A2. Ce gène code pour une protéine située dans les lysosomes, les compartiments cellulaires chargés du traitement des déchets et du recyclage. Les personnes porteuses de mutations d’ATP13A2 peuvent aussi développer une pathologie marquée par des dépôts de fer dans le cerveau. Ce lien fait d’ATP13A2 un point d’entrée idéal pour étudier comment l’équilibre du fer se dérègle. En utilisant une lignée cellulaire humaine de type neuronal surexprimant la protéine de Parkinson alpha‑synucléine, l’équipe a utilisé de petits fragments d’ARN pour réduire l’expression d’ATP13A2, puis a suivi les changements dans le fer, la production d’énergie et la santé cellulaire.
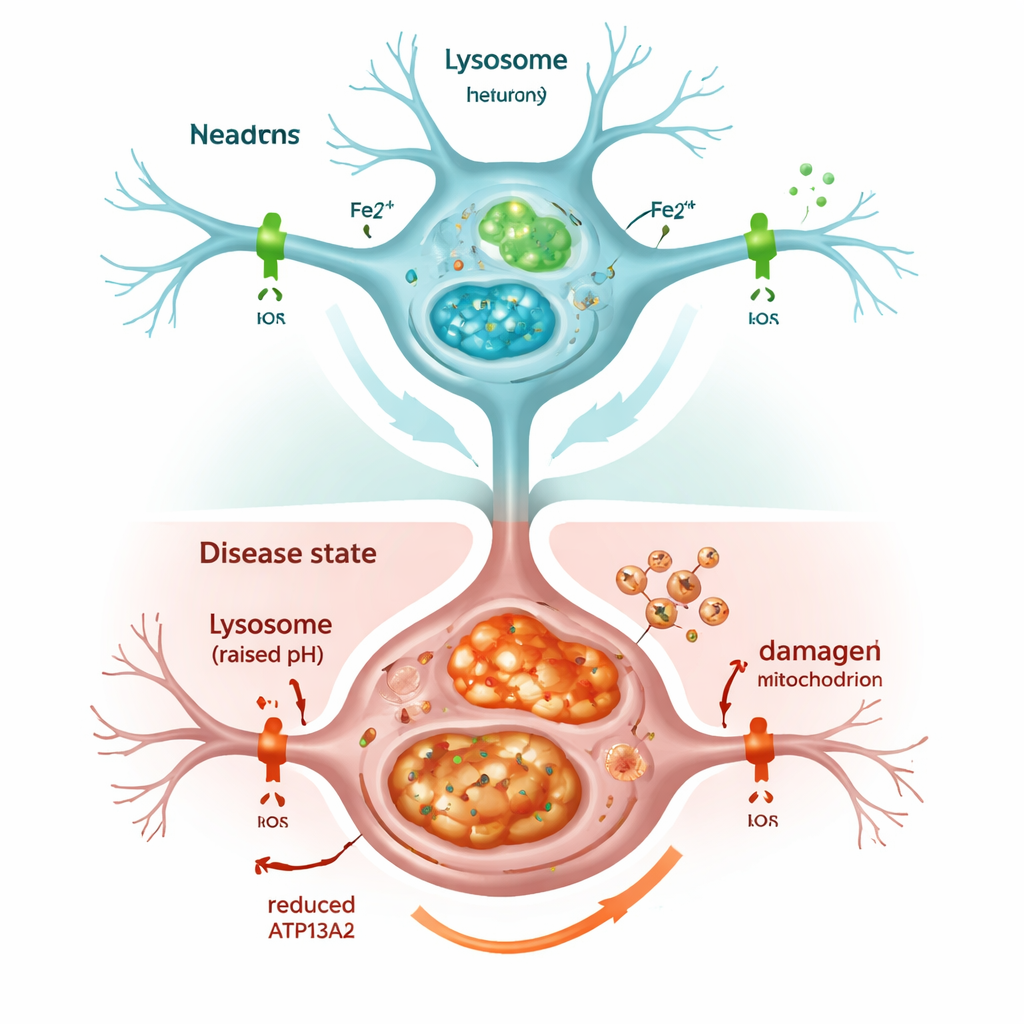
Quand le système de recyclage cellulaire cale
La suppression d’ATP13A2 a rapidement fragilisé les lysosomes. Leur acidité interne, essentielle pour dégrader les matériaux indésirables, a diminué, et les marqueurs du processus d’élimination cellulaire, appelé autophagie, se sont accumulés au lieu d’être éliminés. Par conséquent, l’alpha‑synucléine s’est accumulée, rappelant ce qu’on observe dans les cerveaux atteints de Parkinson. Les cellules présentaient aussi davantage de fer globalement, et en particulier plus de la forme chimiquement active, appelée Fe2+, à l’intérieur des lysosomes et des mitochondries. La cellule a réagi en produisant plus de ferritine, une protéine de stockage du fer, mais cela n’a pas suffi à éviter les dégâts : les mitochondries surchargées ont généré un excès de molécules réactives de l’oxygène, et la survie cellulaire a diminué. Le traitement des cellules par un médicament chélateur du fer, similaire à certains utilisés en clinique, a réduit ce stress oxydatif et partiellement restauré la viabilité cellulaire, soulignant que l’excès de fer lui‑même était un moteur clé des dommages.
Les capteurs du fer cessent d’écouter le métal
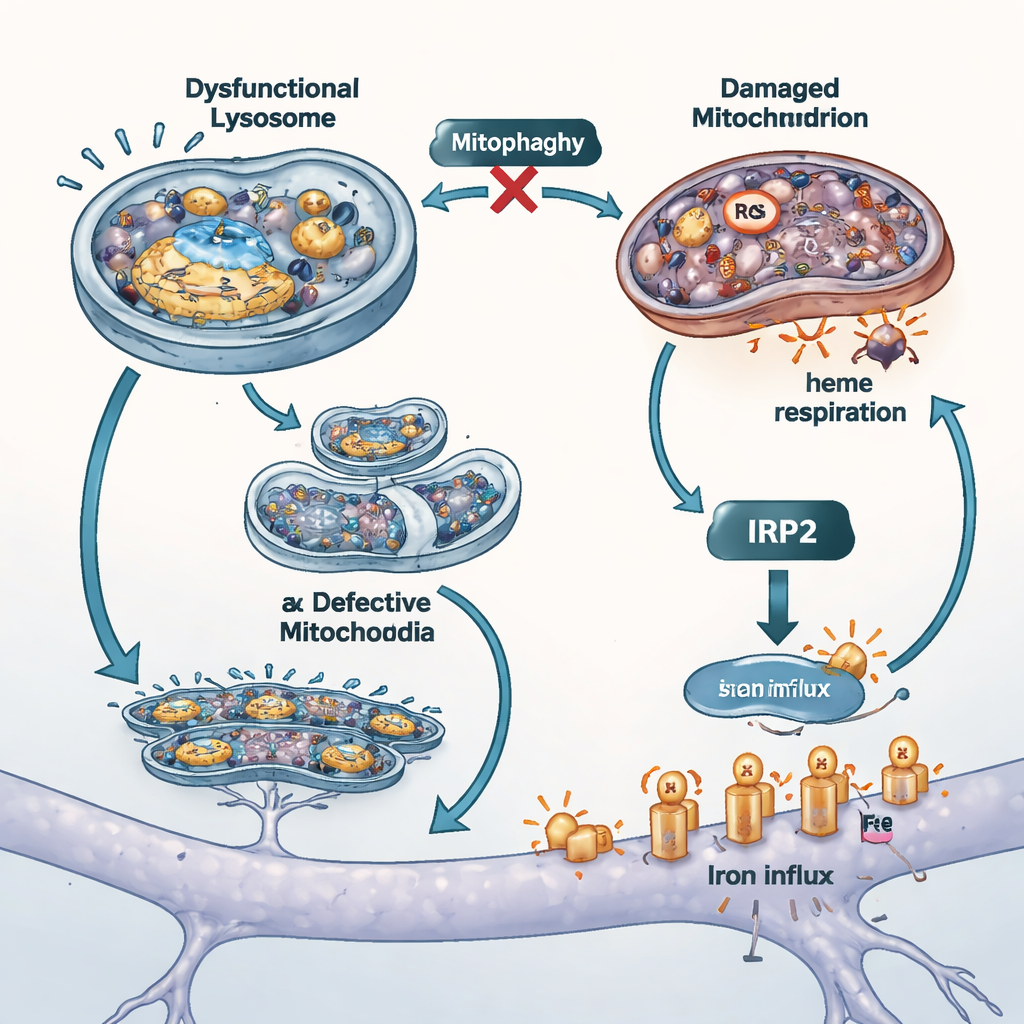
D’ordinaire, les cellules disposent d’un système de rétrocontrôle qui détecte la hausse des niveaux de fer et répond en réduisant l’importation de fer. Une protéine appelée IRP2 détecte le fer, en partie via un signal dépendant de l’hème provenant des mitochondries, puis ajuste la production des protéines transportant le fer à la surface cellulaire. Dans les cellules déficientes en ATP13A2, ce mécanisme de protection a échoué. Les transporteurs qui font entrer le fer dans la cellule sont restés élevés alors même que le fer était déjà augmenté. Les niveaux de protéine IRP2 ont à peine varié, et l’ajout d’un supplément de fer externe n’a pas déclenché sa dégradation habituelle. L’équipe a rattaché cette défaillance aux mitochondries : celles‑ci présentaient une respiration moins efficace, des signes de contrôle qualité défectueux (mitophagie) et, surtout, perdaient la capacité de synthétiser l’hème, la molécule contenant du fer qui aide IRP2 à détecter le fer. Sans suffisamment d’hème, IRP2 ne recevait pas le message « trop de fer » et laissait se poursuivre l’afflux de fer.
Fermer le robinet à fer et tester d’autres modèles
Pour mesurer la contribution de cet apport incontrôlé de fer aux lésions cellulaires, les scientifiques ont bloqué deux voies principales d’entrée du fer. Ils ont utilisé une transferrine dépourvue de fer pour concurrencer un importateur, et un petit médicament pour atténuer l’activité d’un autre transporteur nommé DMT1. Les deux interventions ont réduit la quantité totale et la quantité libre de fer à l’intérieur des cellules, diminué le stress oxydatif mitochondrial et amélioré la survie, suggérant que les canaux d’entrée du fer à la surface sont des amplificateurs importants des dommages lorsque ATP13A2 est perdu. Les chercheurs ont aussi répété des expériences clés dans des cellules dépourvues d’un autre gène lié à la maladie de Parkinson, PINK1, connu pour altérer la mitophagie. Ces cellules ont montré la même combinaison d’accumulation de fer et d’affaiblissement de la production d’hème, soutenant l’idée que le contrôle qualité mitochondrial et l’équilibre du fer sont étroitement liés dans différentes formes de la maladie.
Ce que cela signifie pour la maladie de Parkinson et les traitements futurs
En termes simples, l’étude décrit un cercle vicieux. Quand ATP13A2 est supprimé, les lysosomes n’éliminent plus correctement les composants endommagés, y compris les mitochondries défectueuses. Ces mitochondries affaiblies produisent alors moins d’énergie et moins d’hème, affaiblissant le système de détection du fer de la cellule. Le fer continue d’affluer via les transporteurs de surface, s’accumule dans des compartiments vulnérables et alimente des réactions toxiques qui endommagent encore davantage les mitochondries. Avec le temps, ce boucle peut aider à expliquer pourquoi certains neurones meurent dans la maladie de Parkinson et dans les troubles cérébraux associés à une surcharge en fer. Les conclusions suggèrent que des thérapies futures pourraient non seulement viser à éliminer l’excès de fer, mais aussi restaurer le bon fonctionnement des lysosomes, le contrôle qualité mitochondrial et la production d’hème — attaquant la cause profonde du problème plutôt que de se contenter d’éponger le fer après coup.
Citation: Murakami, T., Ohuchi, K., Kiuchi, M. et al. Disruption of intracellular iron homeostasis through mitochondrial dysfunction associated with suppression of ATP 13A2 expression. Sci Rep 16, 5007 (2026). https://doi.org/10.1038/s41598-026-35368-x
Mots-clés: Maladie de Parkinson, fer cérébral, mitochondries, lysosomes, synthèse de l’hème