Clear Sky Science · fr
Investigation des résidus points chauds fonctionnels d’une enzyme par la surveillance en temps réel de la réaction enzymatique par RMN et approches computationnelles
Pourquoi cela compte pour les futurs antiviraux
Le favipiravir est un comprimé déjà utilisé contre la grippe et testé pour le COVID‑19, mais il n’attaque pas le virus sous la forme que nous avalons. Nos propres cellules doivent d’abord le convertir en une molécule active qui bloque le virus. Cette étude dissèque, presque atome par atome, comment une enzyme humaine réalise une étape d’activation cruciale, et quelles petites régions de l’enzyme servent de « points chauds » contrôlant la vitesse et l’efficacité de l’activation du médicament. Comprendre ces détails pourrait orienter la conception d’antiviraux de nouvelle génération à la fois plus puissants et plus prévisibles chez les patients.
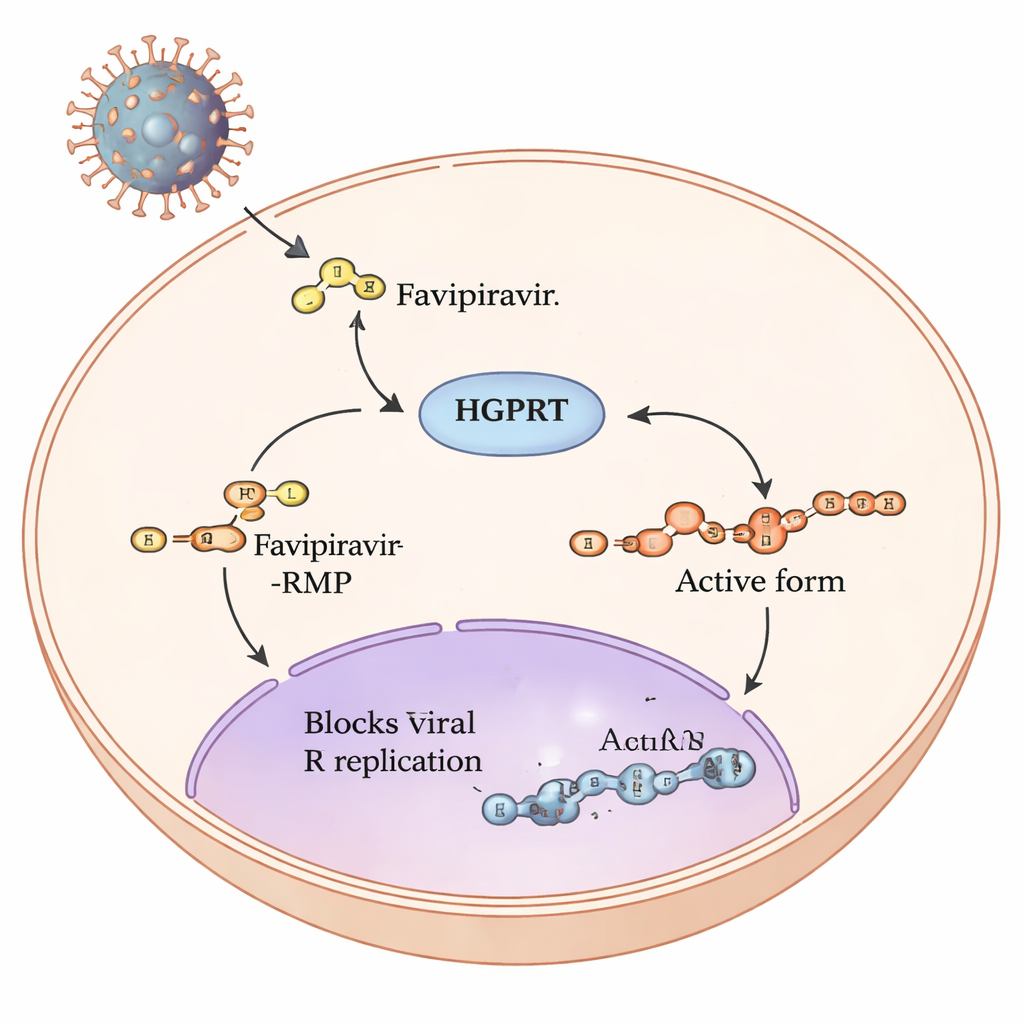
Le parcours d’un prodrogue à l’intérieur de nos cellules
Le favipiravir est un prodrogue : une fois qu’il pénètre dans les cellules humaines, une série d’étapes chimiques le transforme en une forme capable de bloquer la machinerie de réplication des virus à ARN comme la grippe et le SARS‑CoV‑2. La première et la plus lente de ces étapes est assurée par une enzyme humaine appelée hypoxanthine‑guanine phosphoribosyltransférase, ou HGPRT. HGPRT ajoute un petit groupe sucre‑phosphate au favipiravir, produisant le favipiravir‑RMP. Ce n’est qu’après cette étape que d’autres enzymes peuvent assembler la forme triphosphate pleinement active qui interfère directement avec l’ARN polymérase virale. Parce que cette première étape catalysée par HGPRT fait office d’entonnoir limitant la quantité de médicament actif produite, les auteurs ont cherché à identifier quelles parties de HGPRT sont les plus importantes pour traiter le favipiravir.
Observer la chimie en temps réel par RMN
De façon unique, le favipiravir contient un atome de fluor qui se comporte comme un petit émetteur radio dans un champ magnétique. L’équipe a exploité cette propriété en utilisant la spectroscopie RMN du fluor‑19 pour observer, en temps réel, la quantité de favipiravir et de favipiravir‑RMP présentes dans un tube à essai au fur et à mesure de la réaction. Comme seul le médicament porte du fluor, les signaux RMN sont nets et faciles à suivre. En enregistrant des spectres à intervalles pendant 12 heures, les chercheurs ont pu suivre la disparition du composé de départ et l’apparition du produit modifié, puis extraire des mesures cinétiques standard telles que la vitesse de la réaction et l’affinité apparente de l’enzyme pour le médicament.
Modifier des positions clés dans l’enzyme
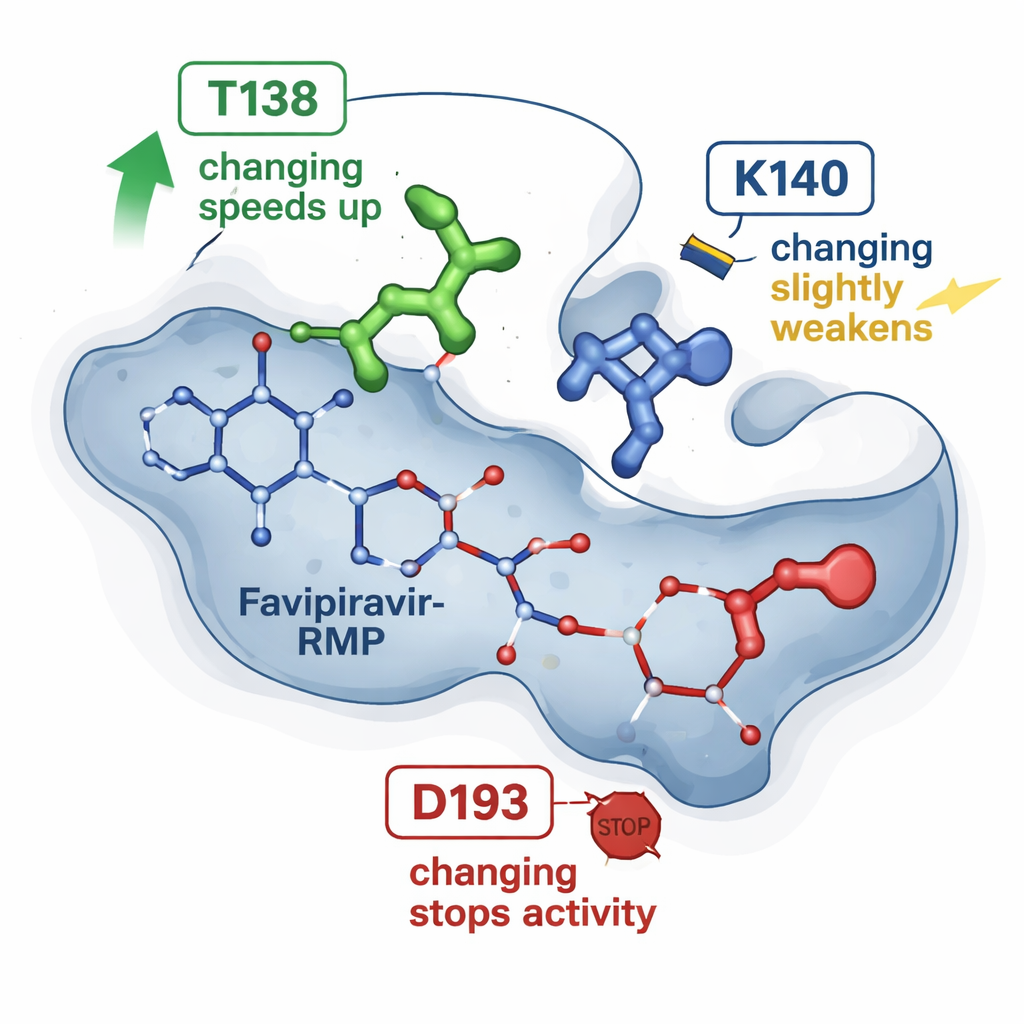
Des clichés antérieurs par cristallographie montrant HGPRT liée au favipiravir‑RMP avaient suggéré une poignée d’acides aminés qui bercent le médicament dans une poche. Le travail nouveau teste trois de ces positions en réalisant des substitutions précises d’une seule lettre dans la protéine et en comparant chaque enzyme mutante à la forme naturelle. Une modification, appelée T138A, a étonnamment rendu l’enzyme environ quatre à six fois plus rapide pour convertir le favipiravir, bien qu’elle ait supprimé un groupe chimique que l’on pensait aider à retenir le médicament. Une deuxième modification, K140M, a légèrement ralenti la réaction et affaibli un peu l’affinité apparente. Une troisième modification, D193N, a complètement supprimé la capacité de l’enzyme à produire le favipiravir‑RMP, même si la protéine altérée pouvait encore être exprimée et lier le produit. Ces résultats montrent qu’ils n’ont pas tous la même importance : certains points de contact agissent comme des régulateurs subtils de vitesse, tandis que d’autres sont des commutateurs essentiels.
Simuler les pièces en mouvement sur ordinateur
Pour aller au‑delà des structures statiques, les chercheurs se sont tournés vers des simulations moléculaires. À partir de la structure tridimensionnelle connue de HGPRT avec le favipiravir‑RMP, ils ont utilisé des outils computationnels établis pour estimer la force d’interaction du médicament dans chaque mutant et pour exécuter de nombreuses courtes simulations de dynamique moléculaire. Ces simulations suivent le ballet des atomes et leurs interactions sur des dizaines de nanosecondes. Les calculs concordent avec les tendances issues de la RMN : la variante T138A tendait à retenir le favipiravir‑RMP de façon plus favorable tout en montrant des épisodes où le médicament se dirigeait vers une voie d’« échappement », guidé par un autre résidu (K140) qui ancre brièvement le groupe phosphate avant sa libération. En revanche, la variante D193N retenait encore le produit, mais échouait probablement à une étape catalytique antérieure requérant un ion magnésium, ce qui explique la perte d’activité malgré une liaison stable.
Une feuille de route pour une conception antivirale plus intelligente
En combinant des mesures RMN en temps réel avec des modèles informatiques détaillés, cette étude cartographie les points chauds fonctionnels de HGPRT qui gouvernent l’efficacité d’activation du favipiravir. Pour un public non spécialiste, la conclusion est que nos propres enzymes peuvent fortement influer sur la quantité de médicament antiviral actif qui s’accumule dans les cellules, et que modifier soit la forme du médicament soit la poche enzymatique peut changer radicalement ce résultat. La stratégie hybride des auteurs offre un schéma général pour étudier comment d’autres médicaments interagissent avec leurs protéines cibles, pouvant accélérer le développement de nouveaux composés antiviraux mieux adaptés au mécanisme d’activation de l’organisme.
Citation: Sugiki, T., Yoshida, T., Tsukamoto, M. et al. Investigation of the functional hot-spot residues of an enzyme by real-time monitoring of the enzymatic reaction using NMR and computational approaches. Sci Rep 16, 5896 (2026). https://doi.org/10.1038/s41598-026-35354-3
Mots-clés: favipiravir, activation antivirale, enzyme HGPRT, spectroscopie RMN, conception de médicaments