Clear Sky Science · fr
LASSO stochastique pour des données génomiques extrêmement haute dimension
Trouver les aiguilles dans la meule de foin génomique
La biologie moderne peut mesurer des dizaines de milliers de gènes simultanément, mais les études sur des patients ne comprennent souvent que quelques centaines de personnes. Cachés dans ce déséquilibre se trouvent de petits ensembles de gènes qui comptent vraiment pour prédire le risque de maladie ou la survie. Cet article présente « LASSO stochastique », une méthode statistique conçue pour découvrir de manière fiable ces gènes clés au sein d'oceans de données génomiques bruyantes, même lorsqu'il y a beaucoup plus de gènes que de patients.
Pourquoi choisir les bons gènes est si difficile
Les chercheurs s'appuient souvent sur des outils comme le LASSO, qui rabotent les effets des gènes peu importants vers zéro tout en conservant les plus informatifs. Les versions classiques du LASSO, toutefois, peinent lorsque le nombre de gènes dépasse de loin le nombre d'échantillons, comme c'est fréquent en génomique du cancer. Le LASSO standard ne peut sélectionner au maximum qu'autant de gènes qu'il y a de patients, et il a tendance à négliger des gènes qui se comportent de manière similaire. Des améliorations antérieures ajoutant des pénalités supplémentaires peuvent gérer une partie de cette corrélation, mais elles risquent aussi de brouiller le sens biologique en forçant des gènes apparentés à agir comme s'ils poussaient tous les résultats dans la même direction.
Construire des échantillons aléatoires plus propres
Une solution prometteuse consiste à ajuster à plusieurs reprises le LASSO sur de nombreux sous-ensembles plus petits et tirés aléatoirement de gènes, puis à combiner les résultats. Pourtant, ces approches de « bootstrap » souffrent encore de trois problèmes : des gènes corrélés peuvent s'annuler mutuellement, de nombreux gènes sont rarement ou jamais échantillonnés, et l'aléa pur rend la sélection finale instable. Le LASSO stochastique s'attaque frontalement à ces problèmes avec un nouveau schéma d'échantillonnage appelé bootstrap basé sur la corrélation. Plutôt que de choisir les gènes au hasard, il favorise délibérément les gènes qui sont moins corrélés avec ceux déjà choisis, produisant des ensembles de gènes plus restreints et beaucoup plus indépendants. Il veille aussi à ce que chaque gène soit utilisé le même nombre de fois au cours des répétitions du bootstrap, de sorte qu'aucun gène ne soit injustement ignoré. 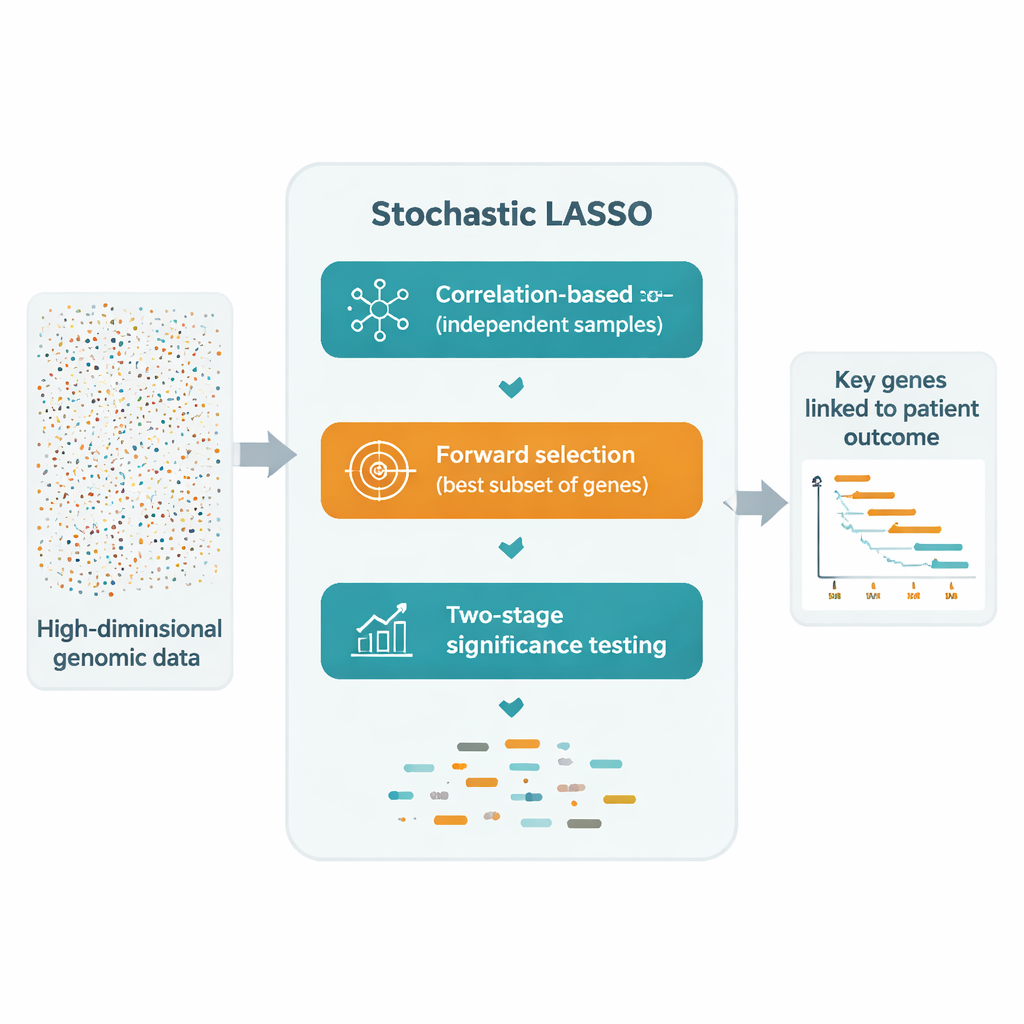
Des indices locaux à un ensemble global de gènes
Après avoir construit ces sous-ensembles plus propres, le LASSO stochastique enregistre l'amplitude du coefficient de chaque gène à travers tous les ajustements bootstrap. Cet effet absolu moyen devient un « score local » qui reflète la constance d'importance d'un gène. Plutôt que de tester exhaustivement chaque combinaison possible, la méthode construit des modèles candidats en ajoutant les gènes par ordre de leurs scores locaux et évalue la qualité de prédiction de chaque candidat sur des données de validation séparées. De cette manière, elle se fixe sur un ensemble compact de gènes dont les signaux combinés expliquent le mieux les données, tout en utilisant bien moins d'essais que les méthodes pas à pas traditionnelles.
Tester quels gènes importent vraiment
Pour passer de « souvent sélectionné » à « statistiquement convaincant », les auteurs introduisent un test t en deux étapes. D'abord, ils vérifient si la moyenne du coefficient de chaque gène à travers les bootstraps est clairement différente de zéro, le signalant comme potentiellement significatif. Ensuite, parmi ces candidats, ils examinent si l'effet de chaque gène est supérieur à la taille d'effet typique de l'ensemble des candidats. Seuls les gènes qui passent les deux tests sont déclarés significatifs. Parce que ces tests s'appuient sur les nombreuses estimations bootstrap, le LASSO stochastique peut identifier avec confiance davantage de gènes significatifs qu'il n'y a de patients — ce que le LASSO conventionnel ne peut pas faire. 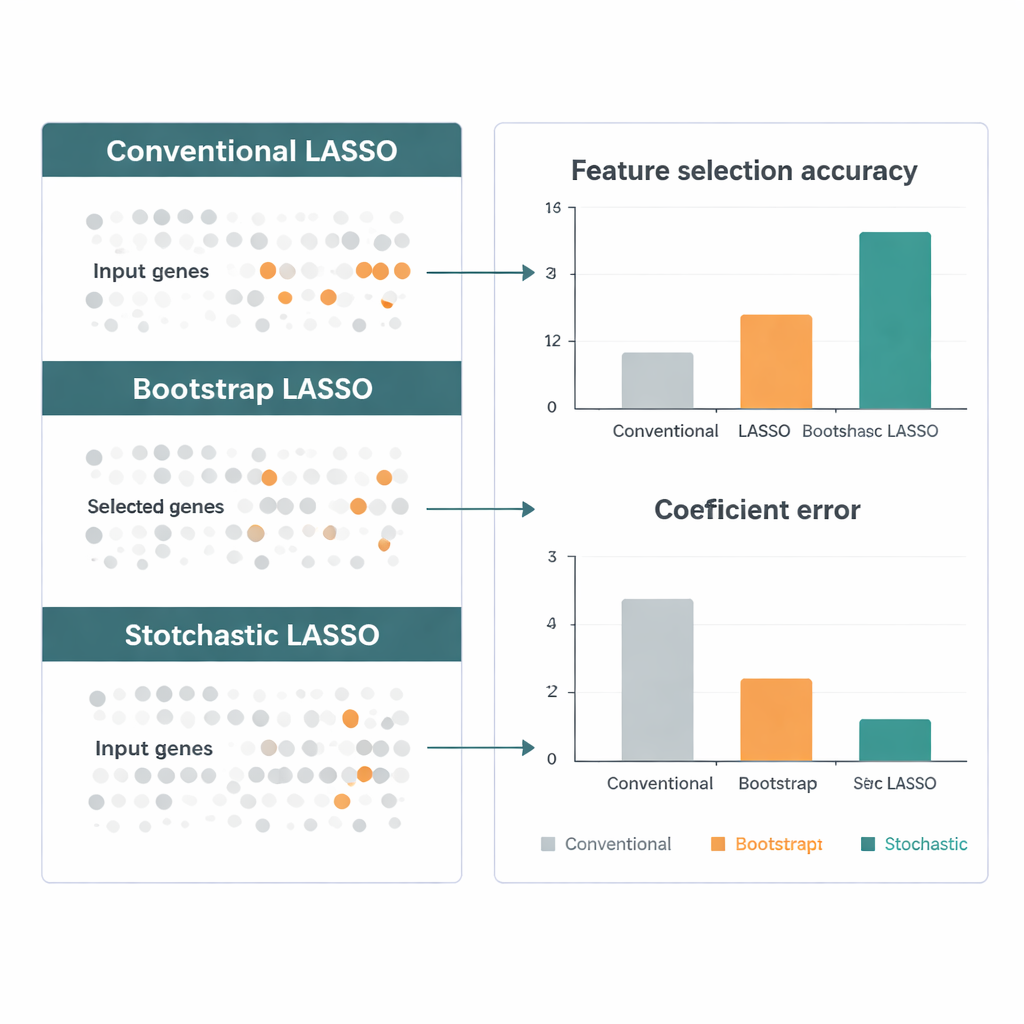
Prouver son utilité dans des simulations et des données de cancer
Les auteurs comparent le LASSO stochastique à plusieurs variantes leaders du LASSO en utilisant des données simulées conçues pour imiter de vraies études génomiques : énormément de gènes, fortes corrélations et signaux « vrais » connus. Dans de multiples scénarios, la nouvelle méthode retrouve les gènes corrects plus souvent, estime leurs effets plus précisément et reste stable d'une exécution à l'autre. Ils se tournent ensuite vers des données d'expression génique du Cancer Genome Atlas pour des tumeurs cérébrales, y compris le glioblastome agressif. Le LASSO stochastique met en évidence des centaines de gènes dont l'activité est liée à la survie des patients et signale des voies biologiques — comme les voies de signalisation et de métabolisme des médicaments — qui bénéficient d'un soutien indépendant dans la littérature, suggérant que la méthode est non seulement statistiquement plus fine mais aussi biologiquement pertinente.
Ce que cela signifie pour les patients et les chercheurs
Pour les non-spécialistes, le message clé est que le LASSO stochastique est un filtre plus intelligent pour les grandes données génomiques. Il aide les scientifiques à séparer les gènes réellement liés à la maladie du bruit statistique, même lorsque les données sont limitées et que les gènes sont fortement interdépendants. En fournissant des listes de gènes et des estimations d'effet plus précises et plus stables, il peut affiner la recherche de biomarqueurs, de cibles thérapeutiques et de signatures pronostiques dans le cancer et d'autres maladies complexes. Bien que démontré sur la régression linéaire, le même cadre peut être intégré aux modèles de survie et aux problèmes de classification, élargissant ainsi son impact potentiel dans la recherche biomédicale.
Citation: Baek, B., Jo, J., Kang, M. et al. Stochastic LASSO for extremely high-dimensional genomic data. Sci Rep 16, 5250 (2026). https://doi.org/10.1038/s41598-026-35273-3
Mots-clés: sélection de caractéristiques génomiques, données haute dimension, méthodes LASSO, expression génique du cancer, découverte de biomarqueurs