Clear Sky Science · fr
Le lactate régule l’axe YTHDF2-FTH1 pour promouvoir la ferroptose des cardiomyocytes et aggraver la lésion d’ischémie-reperfusion myocardique
Pourquoi les patients cardiaques doivent se soucier de cette chimie
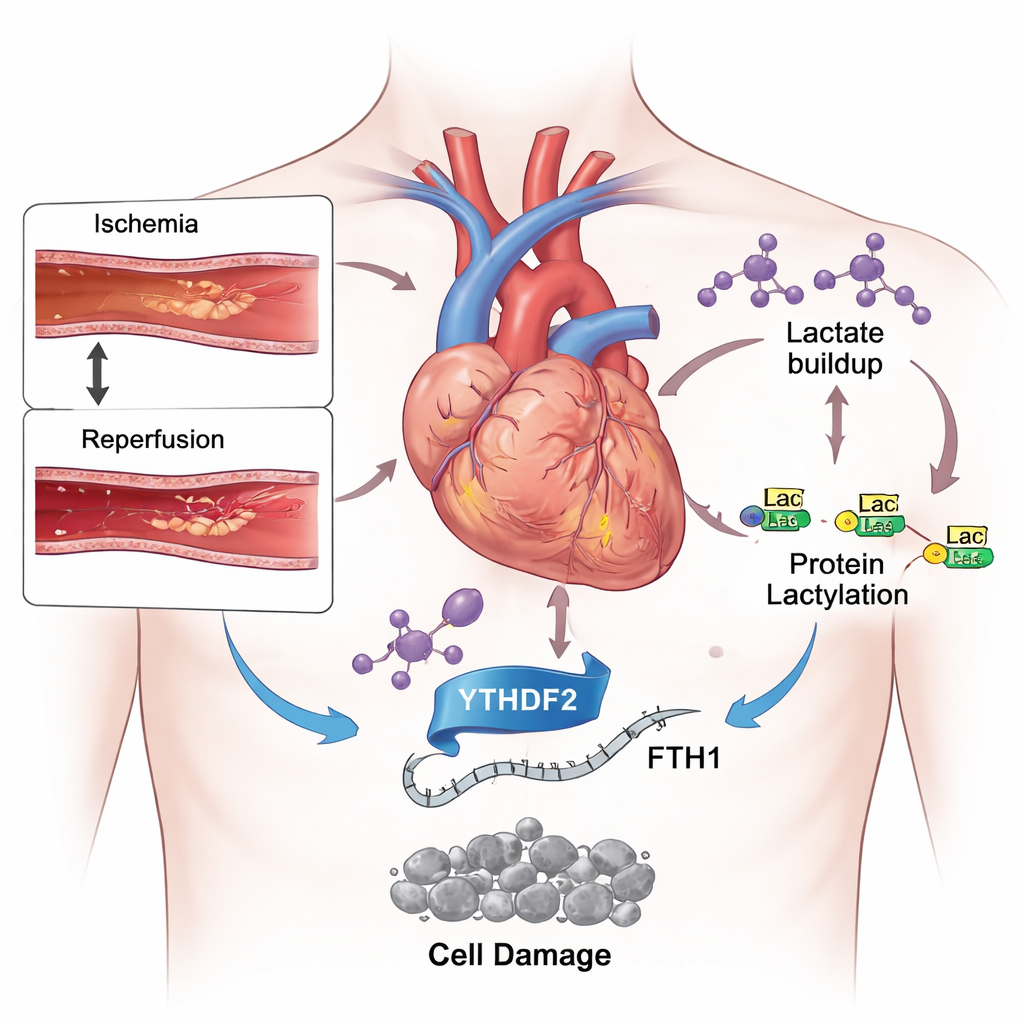
Lorsque les médecins rouvrent une artère coronarienne obstruée après un infarctus, l’arrivée soudaine de sang frais sauve du muscle mais peut aussi causer des dommages supplémentaires, appelés lésion d’ischémie–reperfusion. Cette étude met en lumière un coupable surprenant à l’intérieur des cellules cardiaques : le lactate, un sous-produit métabolique courant. Les auteurs montrent que le lactate peut actionner un commutateur moléculaire qui pousse les cellules cardiaques vers un type particulier de mort cellulaire dépendante du fer, aggravant ainsi la lésion. Comprendre cette voie cachée pourrait pointer vers de nouveaux médicaments protégeant mieux le cœur lors du traitement d’urgence.
Une arme à double tranchant dans le traitement de l’infarctus
La médecine moderne est devenue très efficace pour rouvrir rapidement des artères coronaires obstruées, limitant les dommages initiaux d’un infarctus. Pourtant, les patients peuvent encore perdre de larges zones de muscle cardiaque après le rétablissement du flux sanguin. Une raison est que le retour soudain d’oxygène et de nutriments crée une tempête de stress chimique à l’intérieur des cellules cardiaques. Parmi les différents types de mort cellulaire déclenchés dans ce contexte, une forme plus récente appelée ferroptose a attiré l’attention. Contrairement à des formes plus familières comme l’apoptose, la ferroptose dépend du fer et d’une oxydation incontrôlée des lipides membranaires, ce qui peut affaiblir durablement le cœur.
Comment le lactate devient plus que la simple « douleur musculaire »
Pendant un infarctus, le muscle cardiaque privé d’oxygène bascule son métabolisme vers la glycolyse, un système de secours qui dégrade rapidement le sucre mais produit de grandes quantités de lactate. Chez la souris soumise à un blocage bref puis à la réouverture d’une artère coronaire, et dans des cellules cardiaques cultivées exposées à une hypoxie suivie de réoxygénation, les chercheurs ont observé des niveaux de lactate fortement augmentés. En parallèle, ils ont détecté davantage d’un marquage chimique appelé lactylation sur de nombreuses protéines et sur des histones, les échafaudages organisant l’ADN. Lorsque les animaux ont reçu un médicament ralentissant la glycolyse et réduisant la production de lactate, les lésions cardiaques ont diminué, les marqueurs sanguins de gravité ont baissé, et l’équilibre entre le fer nocif et les antioxydants protecteurs s’est amélioré. Ces résultats suggèrent que l’excès de lactate n’est pas seulement un sous-produit du stress mais un moteur actif des dommages.
Un commutateur moléculaire qui desserre la bride du fer
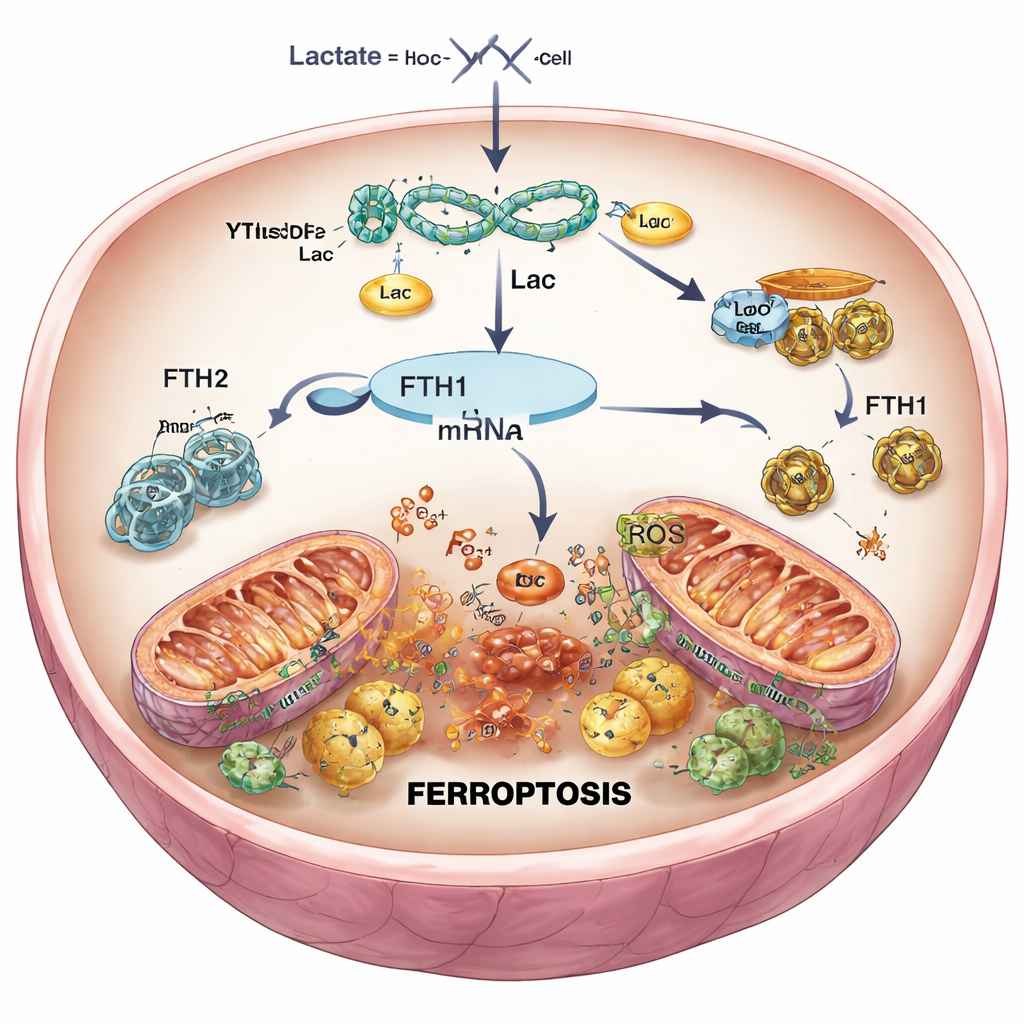
En approfondissant, l’équipe s’est concentrée sur YTHDF2, une protéine qui lit les marques chimiques sur l’ARN et décide de la rapidité avec laquelle certains messages sont détruits. Ils ont découvert que l’ischémie–reperfusion et l’apport de lactate augmentaient tous deux les niveaux de YTHDF2 et accroissaient la lactylation autour du gène qui l’exprime, amplifiant ainsi sa production. L’une des cibles clés de YTHDF2 s’est révélée être l’ARN codant pour la chaîne lourde de la ferritine 1 (FTH1), un élément central de la cage de stockage du fer dans la cellule. FTH1 enferme normalement le fer sous une forme sûre, l’empêchant d’alimenter des réactions dommageables. Dans les cellules cardiaques stressées, YTHDF2 se liait plus fortement à l’ARN de FTH1 et en accélèrait la dégradation, laissant les cellules avec moins de cages ferritine, plus de fer libre, un stress oxydatif accru et des signes caractéristiques de ferroptose.
Atténuer le signal de mort dans les cellules cardiaques
Pour tester la relation de cause à effet, les chercheurs ont utilisé des outils génétiques pour réduire sélectivement YTHDF2 dans les cellules cardiaques et chez la souris. Quand YTHDF2 était inhibé, les niveaux de FTH1 rebondissaient, le fer et les espèces réactives de l’oxygène diminuaient, les mitochondries conservaient une morphologie plus normale, et la survie cellulaire globale s’améliorait après une reperfusion simulée. Chez la souris, une diminution de YTHDF2 se traduisait par des cicatrices d’infarctus plus petites et un tissu d’aspect plus sain. Cependant, lorsque FTH1 était simultanément réduit, ces bénéfices disparaissaient en grande partie : le fer augmentait de nouveau, les dommages oxydatifs revenaient et la taille de l’infarctus augmentait. Cela confirme que YTHDF2 favorise la ferroptose principalement en supprimant FTH1, desserrant ainsi le contrôle du fer à l’intérieur des cellules cardiaques.
Ce que cela signifie pour les thérapies cardiaques futures
En rassemblant les éléments, l’étude décrit une nouvelle chaîne d’événements : une artère bouchée puis rouverte entraîne une accumulation de lactate ; le lactate augmente YTHDF2 via la lactylation ; YTHDF2 détruit ensuite les instructions ARN pour la protéine protectrice du fer FTH1 ; et la surcharge en fer résultante déclenche la ferroptose, approfondissant les lésions cardiaques. Pour les patients, le message est porteur d’espoir : cette voie offre plusieurs nouveaux points d’intervention. Des médicaments qui limitent la signalisation nocive du lactate, bloquent la modification spécifique de YTHDF2 ou préservent la fonction de FTH1 pourraient rendre la reperfusion d’urgence plus sûre et protéger davantage de muscle cardiaque. Bien que ces résultats doivent encore être confirmés sur des tissus humains, ils ouvrent une voie prometteuse vers des traitements plus doux et plus efficaces pour les survivants d’un infarctus.
Citation: Xiang, Z., Xiang, B., Ouyang, T. et al. Lactate regulates the YTHDF2-FTH1 axis to promote cardiomyocyte ferroptosis and aggravate myocardial ischemia-reperfusion injury. Sci Rep 16, 4865 (2026). https://doi.org/10.1038/s41598-026-35130-3
Mots-clés: crise cardiaque, lactate, mort cellulaire dépendante du fer, lésion d’ischémie-reperfusion, protection des cardiomyocytes