Clear Sky Science · fr
Un réseau de neurones graphique informé par la physique pour approximer l’affinité de liaison issue du docking pour DYRK2 dans le repositionnement de médicaments contre la maladie d’Alzheimer
Pourquoi cela importe pour la maladie d’Alzheimer
La maladie d’Alzheimer progresse à l’échelle mondiale, et la plupart des médicaments actuels n’atténuent que les symptômes sans arrêter la maladie. Tester de nouveaux composés en laboratoire est long et coûteux, en particulier pour des protéines cérébrales moins étudiées qui pourraient jouer un rôle dans la mémoire et la santé neuronale. Cette étude explore un raccourci intelligent : utiliser un modèle d’intelligence artificielle conscient des lois physiques pour prédire dans quelle mesure des médicaments existants contre Alzheimer pourraient se lier à une protéine peu étudiée appelée DYRK2, ouvrant potentiellement de nouvelles voies thérapeutiques.
Une nouvelle façon d’examiner de vieux médicaments
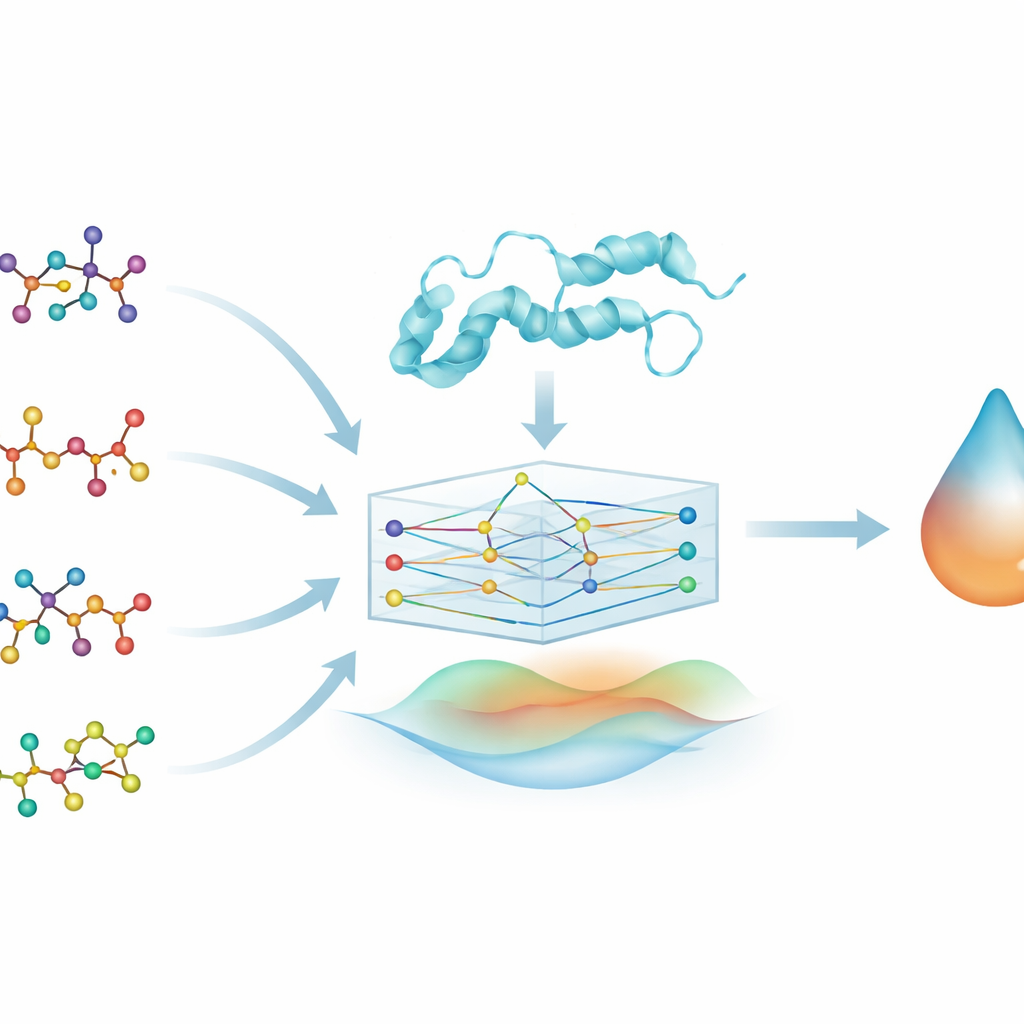
Plutôt que de concevoir de nouveaux composés à partir de zéro, les chercheurs s’intéressent au repositionnement de médicaments : trouver de nouveaux usages pour des médicaments déjà approuvés et dont la sécurité est mieux connue. Ils étudient quatre médicaments familiers contre Alzheimer (brexpiprazole, donépézil, galantamine et rivastigmine) et cherchent à évaluer l’affinité de liaison de chacun pour DYRK2, une kinase impliquée dans la croissance et le fonctionnement des neurones. DYRK2 a été peu étudiée dans le contexte de la maladie d’Alzheimer, mais des preuves initiales la relient aux synapses, aux axones et à la mémoire, ce qui en fait une cible intrigante susceptible de compléter les thérapies actuelles.
Transformer les molécules en réseaux
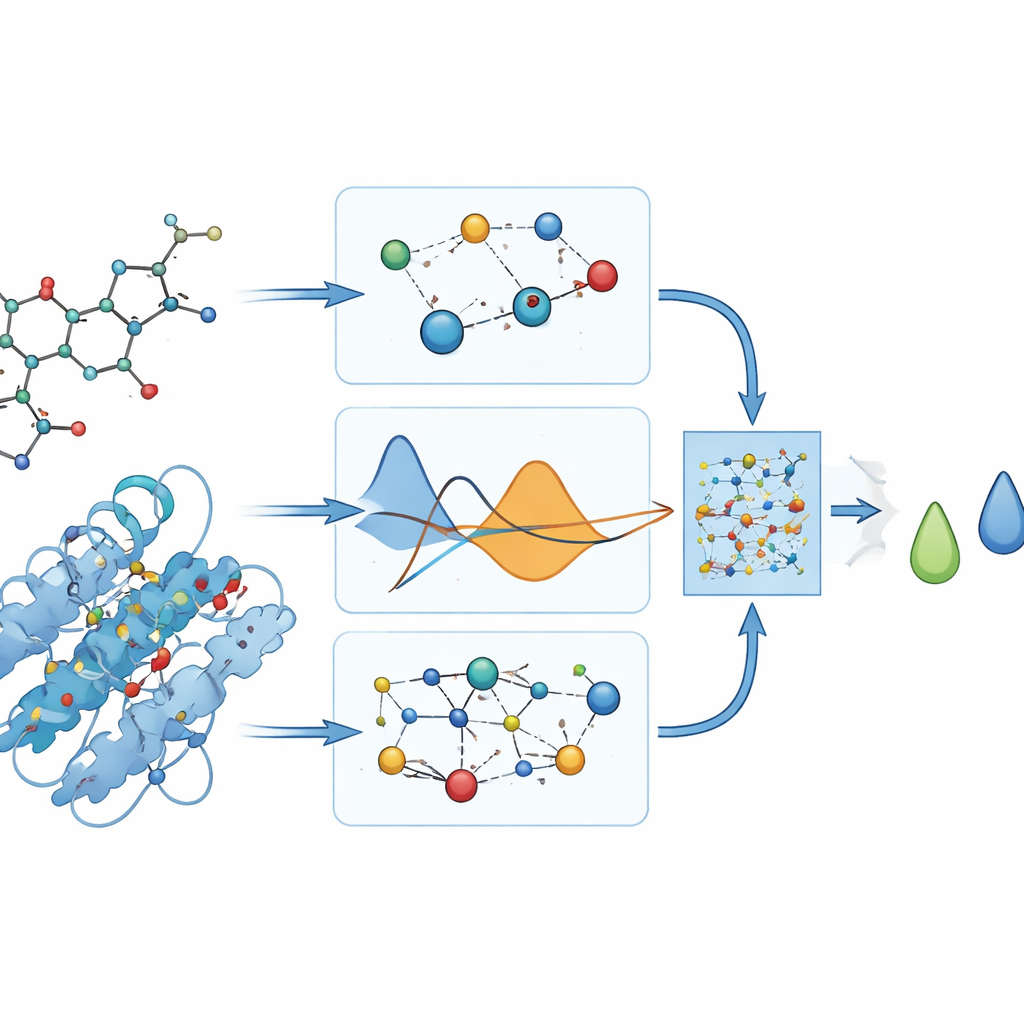
Pour explorer ces relations médicament–protéine, l’équipe convertit chaque molécule en un graphe : les atomes deviennent des nœuds et les liaisons chimiques deviennent des arêtes qui les relient. Ils appliquent une approche similaire au protein DYRK2, représentant sa séquence d’acides aminés comme une chaîne d’unités connectées. Un type de modèle d’apprentissage automatique appelé réseau de neurones graphique (GNN) peut naturellement traiter ces entrées en forme de graphe, en faisant circuler l’information le long des connexions pour apprendre des motifs de forme et de chimie. Cela permet au modèle, nommé PhysDual‑GCN, de « lire » à la fois le médicament et DYRK2 comme des réseaux interagissants plutôt que comme de simples chaînes ou listes de caractéristiques.
Mêler physique et intelligence artificielle
La plupart des outils d’apprentissage profond en découverte de médicaments apprennent uniquement à partir des données, ce qui peut rendre leur fonctionnement interne difficile à interpréter. Ici, les auteurs intègrent délibérément des notions physiques de base sur l’interaction des atomes. Parallèlement aux caractéristiques graphiques apprises, PhysDual‑GCN calcule deux termes énergétiques classiques : l’un capturant l’attraction et la répulsion électriques entre charges partielles, et l’autre décrivant les forces de van der Waals. Ces énergies basées sur la physique sont combinées avec la représentation interne du GNN avant que le modèle n’émette une prédiction d’affinité de liaison. En pratique, le modèle est entraîné à imiter le comportement des programmes de docking classiques — en particulier AutoDock Vina et outils apparentés — mais de manière plus rapide, tout en restant ancré dans des principes physiques familiers.
Ce que le modèle prédit réellement
Étant donné qu’il n’existe pas de mesures expérimentales de la force de liaison de ces médicaments à DYRK2, les auteurs s’appuient sur des programmes de docking pour fournir des scores de liaison « de référence » en unités d’énergie. Ils évitent soigneusement d’utiliser ces scores dans le processus d’entraînement, et s’en servent uniquement après coup pour évaluer la qualité de l’apprentissage de PhysDual‑GCN. Pour les quatre médicaments étudiés, le modèle reproduit les valeurs de docking avec de petites erreurs moyennes (environ un tiers de kilocalorie par mole) et classe correctement les composés : le donépézil et le brexpiprazole ressortent comme les plus forts ligands, tandis que la galantamine et la rivastigmine paraissent plus faibles mais restent relativement stables. Ces résultats montrent que le GNN informé par la physique peut servir de substitut informatique aux runs de docking plus lents.
Promesses et limites de l’approche
Malgré ces chiffres encourageants, les auteurs soulignent les limites strictes de leur étude. Seuls quatre médicaments ont été examinés, et toutes les évaluations reposent sur d’autres programmes informatiques plutôt que sur des expériences biochimiques réelles. La protéine DYRK2 est modélisée principalement comme un graphe unidimensionnel de séquence, et non comme une structure tridimensionnelle complète, de sorte que le modèle ne peut pas encore tenir compte de la géométrie détaillée des poches de liaison. Les énergies physiques elles‑mêmes sont simplifiées, utilisant des paramètres et des coupures standard de champs de force. Par conséquent, ce travail doit être considéré comme une preuve de concept : il montre que des GNN guidés par la physique peuvent suivre de près les scores de docking classiques dans un contexte de peu de données, mais il ne prouve pas encore que les prédictions correspondent à la réalité en laboratoire ou en clinique.
Ce que cela signifie pour la recherche future sur Alzheimer
Pour un public non spécialiste, le message principal est que des algorithmes intelligents et conscients de la physique peuvent aider les scientifiques à explorer de nouvelles cibles pour Alzheimer comme DYRK2 beaucoup plus rapidement que les méthodes traditionnelles seules. En mettant en évidence le donépézil et le brexpiprazole comme des liaisons prometteuses pour DYRK2 et en offrant une manière transparente d’approximer les résultats de docking, PhysDual‑GCN fournit un point de départ pour des études expérimentales plus approfondies. Avec des bibliothèques de médicaments plus vastes, des informations protéiques 3D plus riches et une validation expérimentale, ce type de modèle pourrait devenir un outil pratique pour cribler des candidats thérapeutiques et orienter les efforts de repositionnement visant à ralentir ou modifier le cours de la maladie d’Alzheimer.
Citation: Gider, V., Budak, C. A physics-informed graph neural network to approximate docking-based binding affinity for DYRK2 in Alzheimer’s drug repurposing. Sci Rep 16, 8357 (2026). https://doi.org/10.1038/s41598-026-35102-7
Mots-clés: maladie d’Alzheimer, repositionnement de médicaments, réseaux de neurones graphiques, liaison protéine–ligand, kinase DYRK2