Clear Sky Science · fr
Séquençage métagénomique identifie des agents respiratoires potentiels dans un sous-ensemble d’échantillons de surveillance négatifs en PCR
Pourquoi les germes cachés comptent pour tous
Lorsque vous avez mal à la gorge ou que vous toussez, les médecins s’appuient souvent sur des tests rapides pour rechercher les coupables habituels comme la grippe ou le COVID-19. Mais que se passe-t-il lorsque ces tests indiquent « rien trouvé », alors que vous êtes manifestement malade ? Cette étude lève un coin du voile en utilisant une approche puissante basée sur l’ADN pour chercher des agents que les tests standards manquent, révélant un tableau plus complexe des infections respiratoires et des façons dont nous pourrions les suivre à l’avenir. 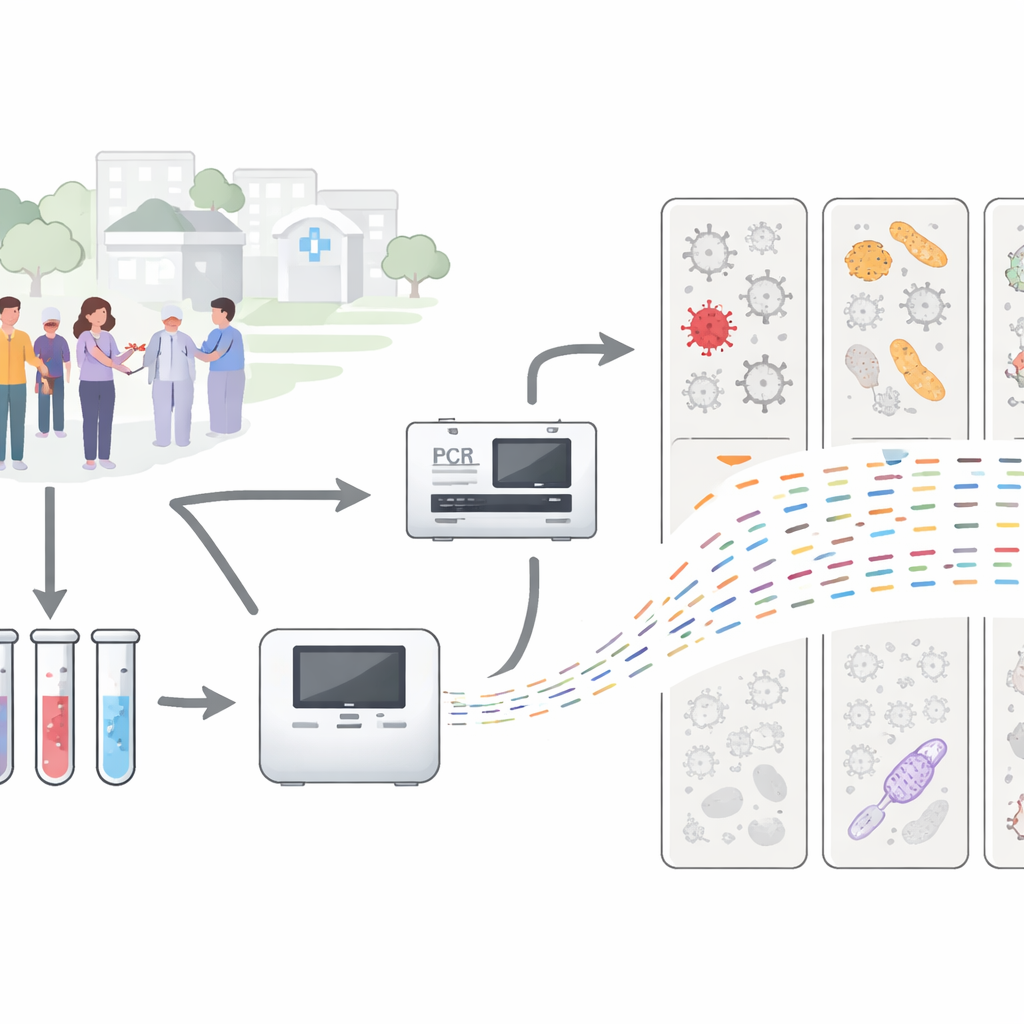
Regarder au‑delà du panel de tests habituel
Pendant la pandémie de COVID‑19, la Californie a mis en place un vaste programme de surveillance des infections respiratoires chez les personnes se rendant en clinique dans plusieurs comtés. L’échantillon nasal ou pharyngé de chaque personne a été testé avec des panels de laboratoire courants qui recherchent une liste fixe de virus et de bactéries, en plus d’un test distinct pour le SARS‑CoV‑2. Plus de la moitié de ces échantillons sont revenus négatifs pour tous les agents de la liste, alors même que les patients présentaient des symptômes nets de rhume ou de grippe. Les chercheurs de cet article ont examiné de plus près 305 de ces échantillons « mystères », ainsi que 26 échantillons déjà connus positifs, pour déterminer si un séquençage plus avancé pouvait révéler ce qui s’y trouvait réellement.
Lire tout le matériel génétique d’un échantillon
Plutôt que de se demander « le virus X est‑il présent ? », l’équipe a utilisé le séquençage métagénomique, qui pose essentiellement la question « quel matériel génétique se trouve dans cet échantillon, quel qu’il soit ? ». Ils ont d’abord extrait tout l’ADN et l’ARN de chaque écouvillon, l’ont copié pour obtenir suffisamment de matériel à analyser, puis l’ont introduit dans des machines de séquençage à haut débit. Dans un sous‑ensemble d’échantillons, ils ont ajouté une étape supplémentaire utilisant un panel de « capture par sondes » conçu pour pêcher le matériel génétique viral, ce qui facilite la détection de virus sinon noyés dans l’abondance de matériel humain ou bactérien. Des programmes informatiques ont ensuite comparé des millions de courts fragments génétiques à d’importantes bases de référence pour déterminer quels virus, bactéries et champignons étaient présents. 
Déceler des virus et microbes négligés
Même parmi les échantillons qui avaient été testés négatifs par les méthodes de routine, l’approche de séquençage a détecté des virus respiratoires humains dans environ 5 % des cas. Il s’agissait notamment du virus influenza C, du bocavirus humain, des rhinovirus et même de quelques infections à SARS‑CoV‑2 que les tests standard avaient manquées. Pour nombre de ces virus, l’équipe a reconstitué des génomes presque complets, ce qui leur a permis d’évaluer la proximité génétique entre souches et avec des virus détectés dans d’autres régions et d’autres années. Ils ont aussi observé que certains échantillons étaient dominés par un type unique de bactérie ou de champignon, comme certaines espèces de Moraxella, Pseudomonas ou Penicillium, suggérant une implication bactérienne ou fongique possible dans la maladie respiratoire ou au moins un rôle dans la structuration de la communauté microbienne locale des voies aériennes.
Ce que les infections manquées peuvent nous apprendre
En reconstruisant des génomes viraux entiers, les chercheurs ont pu dire, par exemple, que les souches de bocavirus dans des comtés voisins étaient presque identiques, ce qui suggère une circulation locale, et que chaque infection à rhinovirus impliquait généralement une souche distincte, y compris une étroitement apparentée à un nouveau type récemment décrit. Ils ont également observé comment l’étape d’enrichissement viral augmentait la quantité et la complétude du matériel génétique viral, en particulier pour les virus plus difficiles à détecter comme influenza C. Dans le même temps, de nombreux échantillons négatifs ne montraient toujours aucun agent clair, soulignant que certains symptômes respiratoires peuvent provenir de causes non infectieuses, d’échantillons de mauvaise qualité ou de microbes présents à des niveaux trop faibles pour être détectés.
Ce que cela signifie pour la surveillance sanitaire future
Pour les soins cliniques quotidiens, les tests ciblés rapides resteront probablement les outils principaux : ils sont moins coûteux, plus rapides et plus simples à exécuter que le séquençage. Mais cette étude montre que lorsque ces tests sont négatifs — en particulier dans les cas graves ou inexpliqués — le séquençage métagénomique large peut révéler des infections cachées, identifier des virus rares ou inhabituels et fournir des génomes complets pour suivre l’évolution des variants au fil du temps. À mesure que la technologie devient plus abordable et standardisée, elle pourrait devenir un complément puissant aux tests de routine, aidant les responsables de santé publique à repérer tôt de nouvelles menaces et à mieux comprendre comment une grande diversité de virus, bactéries et champignons circule dans nos communautés.
Citation: Mascarenhas, A.C., Kantor, R.S., Thissen, J. et al. Metagenomic sequencing identifies potential respiratory pathogens in PCR-negative subset of surveillance samples. Sci Rep 16, 9308 (2026). https://doi.org/10.1038/s41598-025-33917-4
Mots-clés: infections respiratoires, séquençage métagénomique, surveillance virale, tests diagnostiques, découverte de pathogènes