Clear Sky Science · fr
Jeu de données virtuel minimal pour une assemblée de novo triploïde reproductible
Pourquoi les génomes à trois copies comptent
De nombreuses cultures et autres organismes ne portent pas seulement deux copies de chaque chromosome, comme les humains : ils peuvent en porter trois ou davantage. Reconstituer ces copies supplémentaires à partir de données de séquençage est étonnamment difficile, car les copies sont très semblables mais pas strictement identiques. Cet article présente un petit jeu de données « virtuel » soigneusement conçu qui permet aux chercheurs de tester et comparer des logiciels d’assemblage de génomes sur un problème réaliste à trois copies (triploïde), dans des conditions entièrement connues et reproductibles.
Construire un génome de remplacement simple
Plutôt que de partir d’une plante ou d’un animal réel, l’auteur crée d’abord un fragment d’ADN aléatoire d’un million de lettres pour servir de modèle propre. Ce modèle est ensuite dupliqué en trois versions distinctes, représentant les trois jeux de chromosomes d’un organisme triploïde. Pour imiter la manière dont les génomes réels changent lentement au fil du temps, l’étude introduit un nombre fixe de petites modifications — des substitutions d’une seule lettre — étape par étape dans chaque copie. La répétition de ce processus sur 100 étapes produit des triplets de génomes allant de presque identiques à nettement mais modérément différents. Ce « gradient de divergence » contrôlé constitue la colonne vertébrale du banc d’essai.
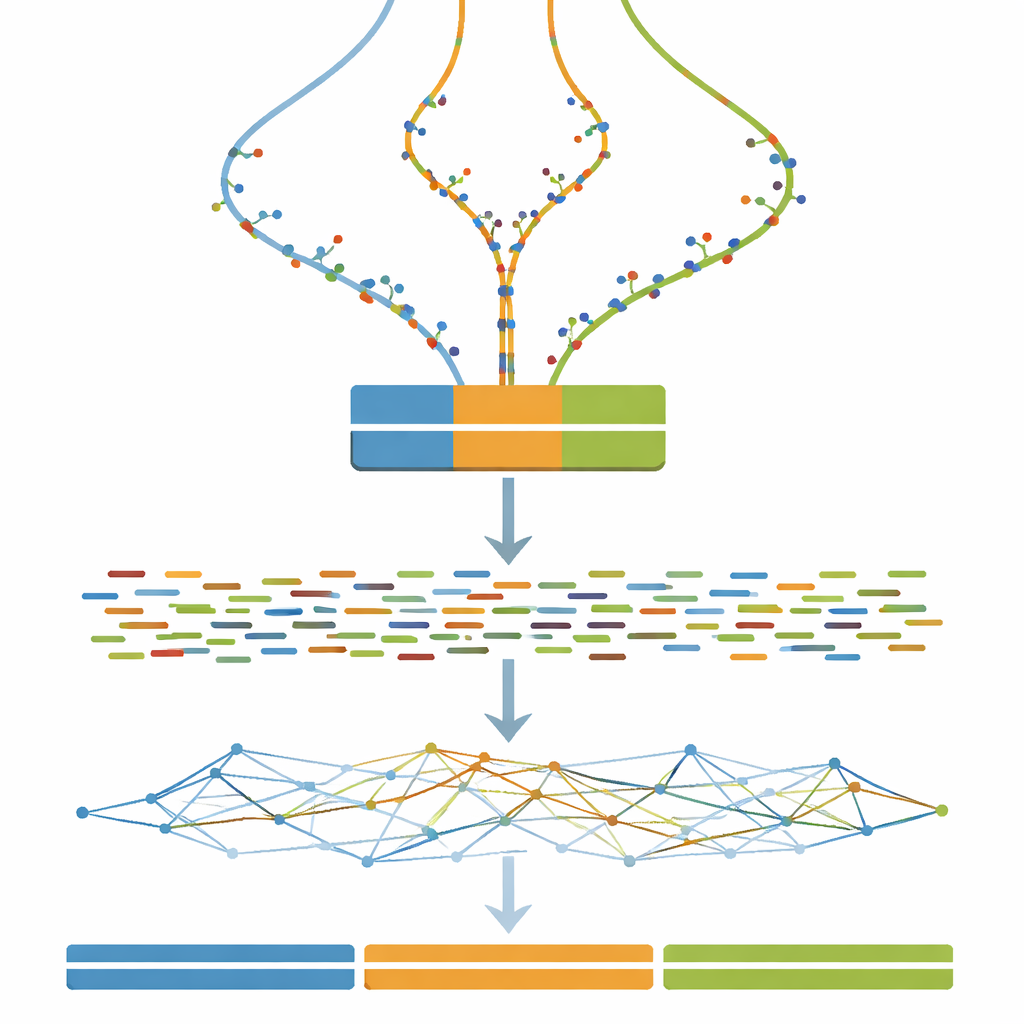
Transformer les génomes virtuels en expériences virtuelles
Une fois chaque génome à trois copies défini, l’étape suivante consiste à imiter ce qu’un appareil de séquençage verrait. L’étude utilise des logiciels largement adoptés pour simuler de courts fragments d’ADN appariés, proches de ceux produits par un séquenceur Illumina, à une profondeur de couverture constante et assez élevée. Des étapes optionnelles de nettoyage reproduisent des pratiques courantes du monde réel, comme la correction d’erreurs de séquençage aléatoires et la fusion de paires de lectures qui se chevauchent. En conséquence, quiconque utilise le jeu de données peut tester non seulement ses algorithmes d’assemblage, mais aussi la manière dont les choix habituels de prétraitement influencent les génomes assemblés finaux.
Soumettre les stratégies d’assemblage à rude épreuve
Le cœur du travail est une vaste expérience dans laquelle toutes les lectures simulées sont fournies à un seul programme d’assemblage de génome en ne changeant qu’un paramètre clé : la taille du k-mer, un réglage qui contrôle le niveau de « découpage » des lectures lorsque le logiciel reconstruit le génome. Pour chaque combinaison de niveau de divergence (de 0 à 100 étapes) et de taille de k-mer (une large plage de valeurs impaires), une nouvelle assemblée est réalisée. Un outil d’évaluation compagnon mesure ensuite la continuité des fragments assemblés, leur nombre, et la proximité de leur longueur combinée avec la vérité connue de trois millions de lettres. Ces mesures sont résumées sous forme de cartes thermiques, révélant de larges zones où les assemblages fusionnent plusieurs copies en une seule, se fragmentent en nombreux petits morceaux, ou s’approchent de l’idéal de trois contigs longs et précis.
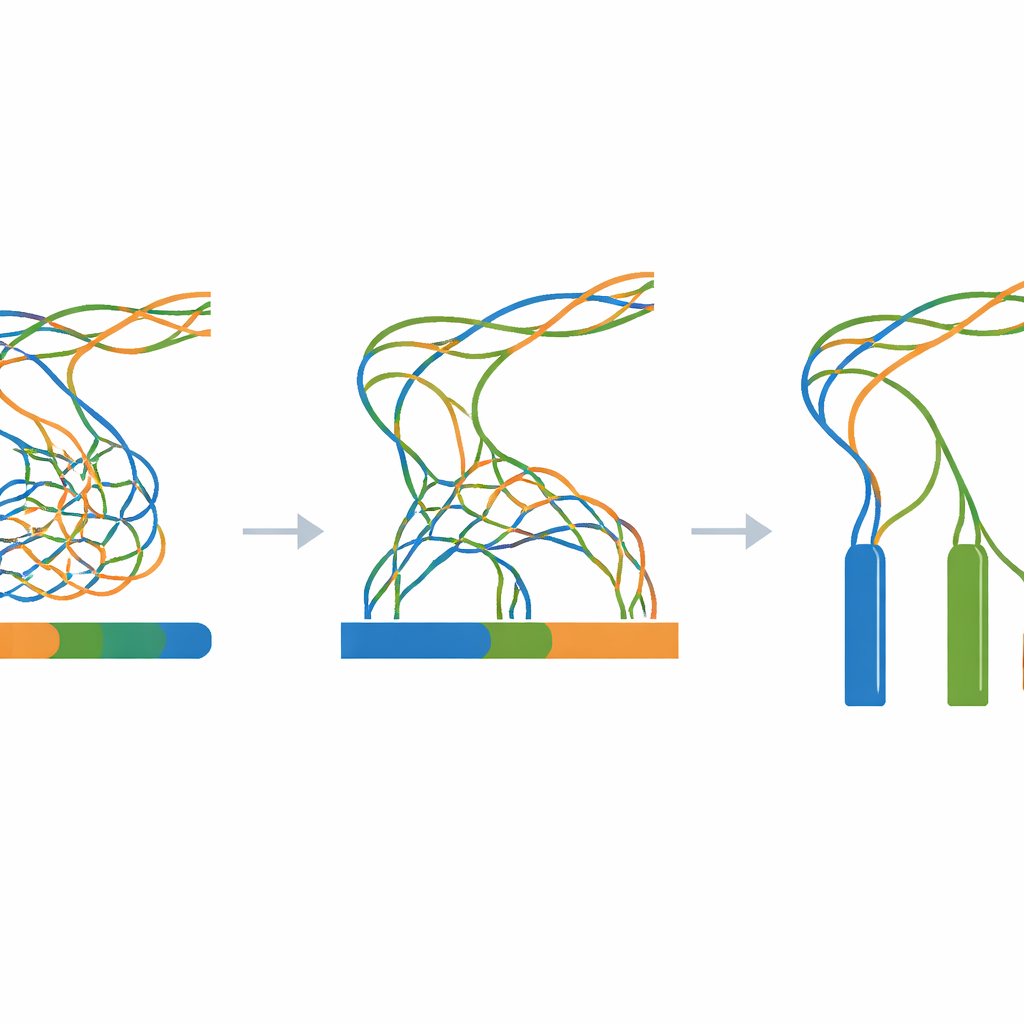
Une référence transparente pour des génomes délicats
Parce que chaque étape est synthétique et scriptée — du modèle aléatoire initial jusqu’aux assemblages finaux — les chercheurs peuvent reproduire l’intégralité du flux de travail sur n’importe quel ordinateur Linux standard en n’utilisant que des outils open source. L’archive Zenodo liée dans l’article contient le génome modèle, toutes les séquences mutées intermédiaires, toutes les lectures simulées et chaque résultat d’assemblage, ainsi que les logs et des scripts d’aide simples. Des vérifications techniques confirment que le processus de mutation se comporte comme prévu, que les lectures simulées correspondent aux longueurs et à la couverture demandées, et que les assemblages montrent le schéma attendu : fort effondrement lorsque les trois copies sont presque identiques, et séparation plus nette à mesure qu’elles divergent davantage.
Ce que cela signifie en termes simples
En langage courant, cet article propose une piste d’essai contrôlée pour les logiciels qui cherchent à reconstruire trois livres d’instructions similaires à partir de tas de fragments mélangés. En augmentant progressivement la différence entre les trois livres, et en modifiant systématiquement un paramètre clé du processus de reconstruction, le jeu de données permet de voir facilement quand et comment les méthodes actuelles échouent ou réussissent. Les développeurs peuvent l’utiliser pour ajuster de nouveaux algorithmes, tandis que les utilisateurs peuvent mieux comprendre quels réglages fonctionnent le mieux pour des génomes triploïdes. Bien que l’ADN lui‑même soit artificiel, les enseignements qu’il permet — sur l’effondrement, la séparation et l’impact des choix de paramètres — sont directement pertinents pour les efforts réels de décodage des génomes complexes de nombreuses espèces importantes.
Citation: Ootsuki, R. Minimum virtual dataset for reproducible triploid de novo genome assembly. Sci Data 13, 382 (2026). https://doi.org/10.1038/s41597-026-06779-2
Mots-clés: assemblage de génome triploïde, évaluation des polyploïdes, jeu de données d’ADN synthétique, assemblage de novo, optimisation de k-mer