Clear Sky Science · fr
Éviter que les données de protéomique ne deviennent des tombes numériques grâce à la responsabilité collective et à l’engagement communautaire
Pourquoi vos données médicales ne devraient pas finir dans un cimetière numérique
La médecine moderne s’appuie de plus en plus sur d’immenses jeux de données décrivant les milliers de protéines à l’œuvre dans nos cellules. Ces fichiers sont souvent partagés en ligne de façon ouverte, promettant que d’autres scientifiques pourront vérifier les résultats ou poser de nouvelles questions sans refaire les expériences. Mais si les données sont publiées dans des formats confus, sans détails clés ou liées à des logiciels propriétaires, elles deviennent des « tombes de données » : visibles par tous, mais pratiquement inutilisables. Cet article montre comment un cours universitaire a transformé des étudiants en détectives des données pour révéler ce problème caché — et propose des solutions simples qui pourraient rendre les données partagées réellement réutilisables.
Apprendre la science en refaisant de vraies études
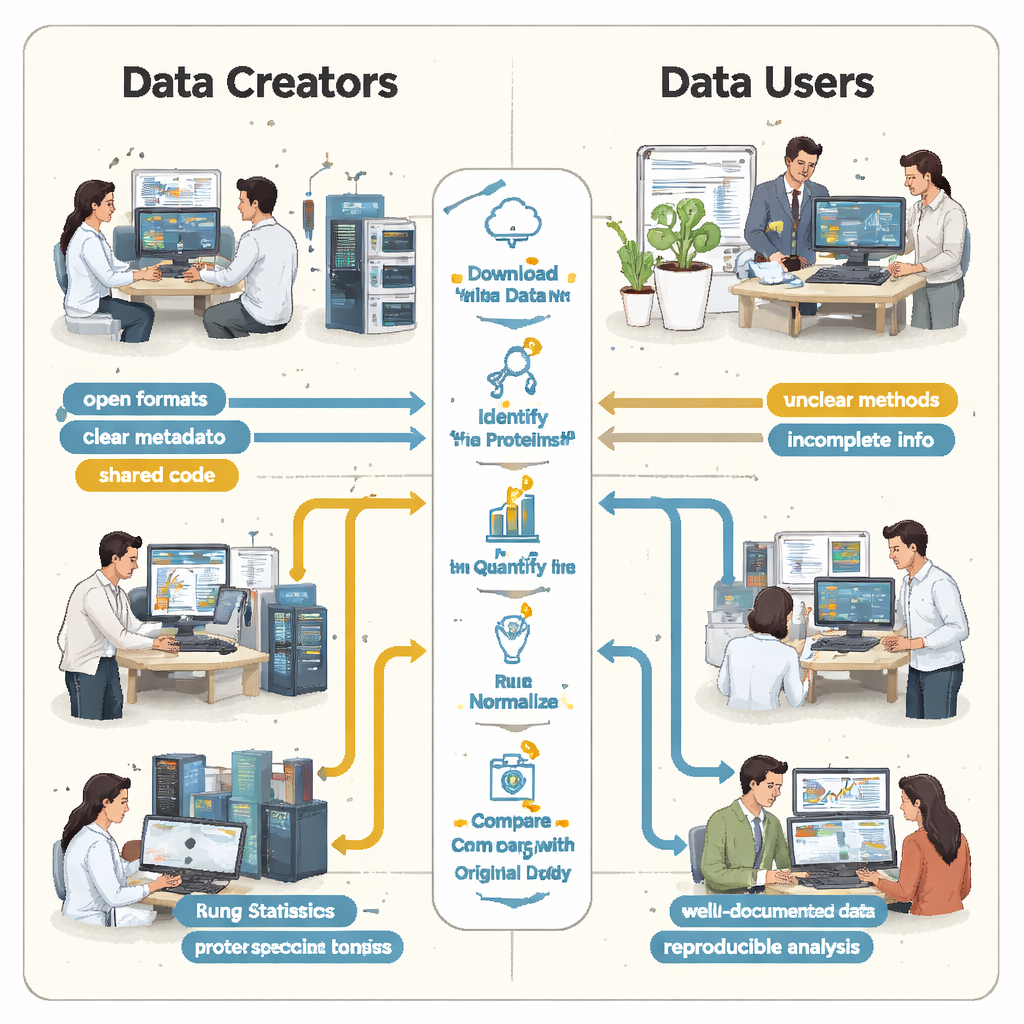
À l’Université d’Helsinki, des étudiants en master suivant un cours de protéomique par spectrométrie de masse ont eu pour mission ambitieuse de choisir de vrais jeux de données protéiques publics issus d’un grand dépôt et d’essayer de reproduire les résultats publiés. Travaillant en petites équipes, ils ont téléchargé six projets depuis le réseau ProteomeXchange, qui héberge des résultats de spectrométrie de masse provenant de nombreux laboratoires à travers le monde. À l’aide d’un pipeline d’analyse partagé en langage R, les étudiants ont suivi les mêmes grandes étapes que les chercheurs originaux : identifier les protéines, mesurer leur abondance, nettoyer les données et tester quelles protéines varient entre des conditions telles que maladie versus tissu sain.
Grandes promesses, instructions manquantes
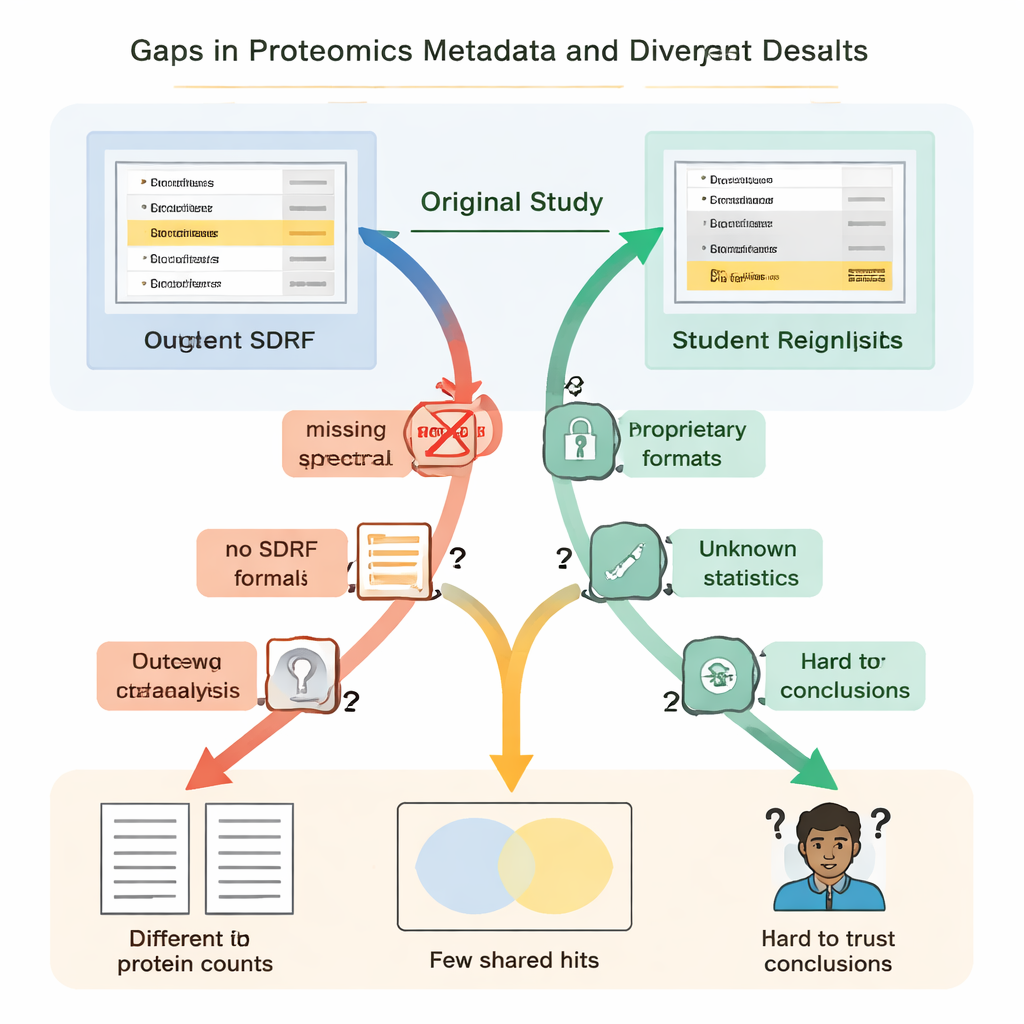
Les étudiants ont vite découvert que « ouvert » ne signifiait pas toujours « réutilisable ». Dans tous les cas, des instructions essentielles étaient absentes ou difficiles à trouver. Les liens clés entre échantillons et fichiers de données n’étaient pas décrits dans un format simple et lisible par machine, si bien que les équipes ont dû deviner quels fichiers bruts correspondaient à quels groupes biologiques en lisant les articles et en déchiffrant les noms de fichiers. Les détails sur la manière dont les faux positifs étaient contrôlés — comme l’utilisation de séquences protéiques « leurres » (decoys) — faisaient défaut, rendant impossible une évaluation rigoureuse de la fiabilité des listes de protéines rapportées. Dans plusieurs projets, les résultats principaux étaient enfermés dans des formats propriétaires ou dépendaient de logiciels commerciaux auxquels les étudiants n’avaient pas accès, les contraignant à reprendre une bonne partie de l’analyse depuis zéro.
Quand de petits manques créent de grandes différences
Ces pièces manquantes n’étaient pas de simples désagréments ; elles ont conduit à des résultats scientifiques radicalement différents. Dans une étude sur une maladie rénale, les auteurs originaux rapportaient un peu moins de cinq mille protéines, tandis que la réanalyse des étudiants — utilisant un outil ouvert et une bibliothèque spectrale construite localement — en a trouvé plus de treize mille. Une protéine mise en avant dans l’article original comme particulièrement importante n’apparaissait pas de façon convaincante dans le fichier d’identification sous-jacent et n’a pas du tout été détectée dans le workflow des étudiants. Dans un autre cas, l’étude originale listait 108 protéines comme variables entre conditions, mais les étudiants, travaillant sur les mêmes données brutes mais sans informations complètes sur les statistiques originales, n’ont pu valider en toute confiance que 11 protéines. Ailleurs, l’absence de réplicats biologiques dans les fichiers déposés rendait tout simplement impossible un test statistique approprié.
Ce qu’un jeu de données « réutilisable » devrait vraiment contenir
De ces six études de cas a émergé un schéma clair : les principaux obstacles à la reproductibilité n’étaient pas les appareils de spectrométrie de masse eux‑mêmes, mais la façon dont les résultats étaient emballés et partagés. Les auteurs soutiennent que chaque jeu de données de protéomique devrait être accompagné d’un paquet minimal de réanalyse. Celui‑ci comprend les données brutes ainsi que des formats de résultats ouverts et standardisés par la communauté ; un tableau normalisé liant chaque échantillon à ses conditions expérimentales ; des résumés de contrôle qualité de base ; toutes les bibliothèques spectrales ou fichiers de séquences protéiques nécessaires pour répéter la recherche ; et les paramètres et le code d’analyse complets, idéalement stockés avec des conteneurs logiciels versionnés. Les dépôts, revues et évaluateurs pourraient aider en incitant ou en exigeant que les déposants fournissent ce paquet dès le départ, afin que d’autres n’aient pas à reconstruire le flux de travail à partir d’indices épars.
Former des scientifiques tout en réparant le système
Le cours lui‑même a servi un double objectif. Pour les étudiants, il offrait une façon pratique de maîtriser des méthodes protéomiques complexes, la statistique et le codage, tout en révélant la fragilité des conclusions publiées lorsque la documentation est incomplète. Pour la communauté au sens large, les difficultés rencontrées par les étudiants ont constitué un test de résistance des pratiques actuelles de partage des données, mettant en lumière les points précis où les métadonnées et les enregistrements d’analyse sont insuffisants. Les auteurs suggèrent que des cours similaires pourraient être organisés ailleurs, transformant les salles de classe en moteurs de contrôle qualité qui poussent en continu vers des données plus claires et plus transparentes.
Des tombes de données à des ressources vivantes
En termes simples, l’article conclut que de nombreux jeux de données protéiques désormais stockés dans des dépôts publics risquent de devenir des cimetières numériques : des expériences coûteuses dont les résultats ne peuvent pas être vérifiés ou étendus de façon fiable. Pourtant la solution est relativement simple : considérer les métadonnées, les formats ouverts et le code partageable comme des parties intégrantes de l’expérience, et non comme des après‑pensées. Si chercheurs, évaluateurs et dépôts insistent collectivement pour un paquet simple et bien documenté à chaque partage de données protéomiques, ces ensembles pourront rester « vivants » : prêts à être réanalysés, combinés avec de nouvelles études et utilisés pour renforcer les preuves à l’appui des découvertes biomédicales.
Citation: Vadadokhau, U., Soliman, M., Castillon, L. et al. Preventing Proteomics Data Tombs Through Collective Responsibility and Community Engagement. Sci Data 13, 287 (2026). https://doi.org/10.1038/s41597-026-06614-8
Mots-clés: protéomique, reproductibilité des données, science ouverte, spectrométrie de masse, partage des données de recherche