Clear Sky Science · fr
AF2BIND : prédire les sites de liaison aux petites molécules en utilisant la représentation par paires d’AlphaFold2
Trouver des cibles médicamenteuses dans un océan de protéines
Les médicaments modernes agissent souvent en se fixant sur de petites anfractuosités à la surface des protéines de nos cellules. Pourtant, malgré les gigantesques catalogues actuels de structures protéiques, il reste étonnamment difficile de prédire à l’avance où une petite molécule — un médicament potentiel — pourrait réellement s’accrocher. Cette étude présente AF2BIND, un outil computationnel simple mais puissant qui exploite le fonctionnement interne d’AlphaFold2, le prédicteur de structures protéiques majeur, pour mettre en évidence des sites de liaison probables à travers des milliers de protéines humaines. Son objectif est de restreindre la recherche de nouveaux médicaments et de révéler des points chauds fonctionnels cachés que les méthodes traditionnelles négligent.
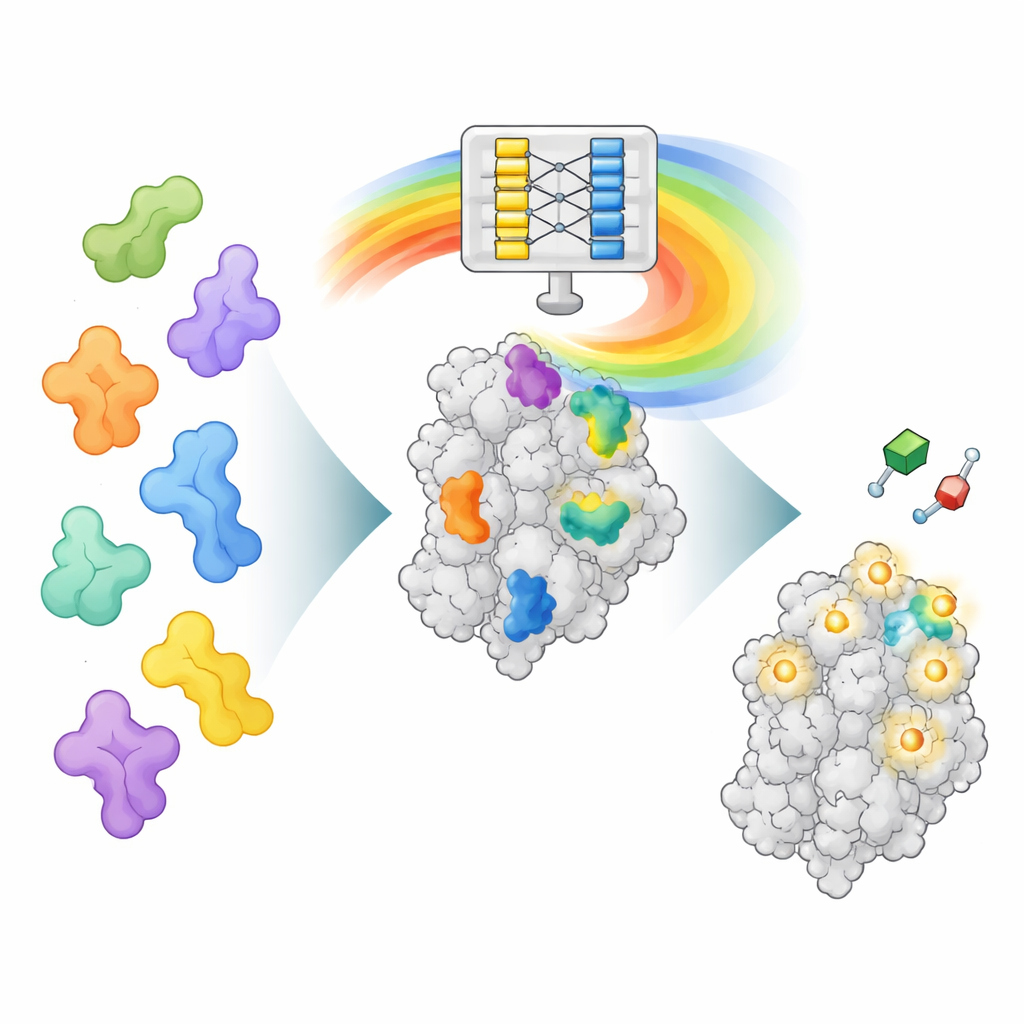
Une nouvelle manière de lire le « cerveau » d’AlphaFold
AlphaFold2 a été entraîné pour prédire comment une chaîne d’acides aminés se replie en une protéine tridimensionnelle, pas pour trouver les sites de liaison des médicaments. Cependant, en apprenant le repliement, il a aussi acquis des motifs riches sur la façon dont différentes parties d’une protéine interagissent. AF2BIND exploite l’une de ces couches de données internes, appelée la représentation par paires, qui encode la relation spatiale entre chaque paire de positions d’acides aminés. Les auteurs fournissent à AlphaFold2 la séquence d’une protéine ainsi que sa structure de squelette et ajoutent également 20 acides aminés supplémentaires, un de chaque type, en tant que chaînes « appât » séparées. AlphaFold2 calcule alors comment la protéine interagit avec chaque résidu appât. Ces patrons d’interaction servent d’entrée à un modèle de régression logistique très simple qui estime, pour chaque position de la protéine, la probabilité qu’elle fasse partie d’un site de liaison à une petite molécule.
Transformer des signaux cachés en prédictions pratiques
L’entraînement d’AF2BIND a nécessité un jeu de données soigneusement sélectionné d’environ 1 900 structures protéine–ligand où les petites molécules étaient liées avec des preuves expérimentales de haute qualité. Les chercheurs ont pris grand soin d’éviter tout « triche » par similarité : ils ont scindé leurs données de sorte que les protéines de test ne partageaient ni repli global, ni séquence, ni même forme de poche de liaison avec celles utilisées pour l’entraînement. Sur ce référentiel rigoureux, la représentation par paires d’AF2 a surpassé plusieurs autres embeddings issus de réseaux neuronaux, y compris ceux basés uniquement sur la séquence ou sur la conception de séquence conditionnée par la structure. En n’utilisant que les caractéristiques par paires, AF2BIND a retrouvé environ les deux tiers des résidus de liaison connus parmi les prédictions les mieux classées et a montré de bonnes performances selon les métriques de classification standard, tout en restant robuste aux modestes variations de forme de la protéine et d’orientations des chaînes latérales.
Lire des indices chimiques à partir des résidus appât
Parce qu’AF2BIND est un modèle linéaire simple, ses décisions sont exceptionnellement transparentes pour un système d’IA moderne. Chacun des 20 acides aminés appât contribue pour une quantité mesurable au score final de liaison en un point donné de la protéine. En examinant ces contributions à travers environ 2 000 complexes protéine–ligand, les auteurs ont constaté que certaines combinaisons d’appâts s’activent plus fortement pour des ligands apolaires, riches en carbone, tandis que d’autres s’éclairent pour des molécules plus polaires et hydrophiles. Autrement dit, le schéma d’activation des appâts agit comme une empreinte chimique grossière indiquant quels types de petites molécules une poche donnée préfère. Cela suggère qu’à l’avenir, des approches de type AF2BIND pourraient non seulement repérer où un médicament pourrait se lier, mais aussi donner des indices sur le type de chimie qui conviendrait le mieux.
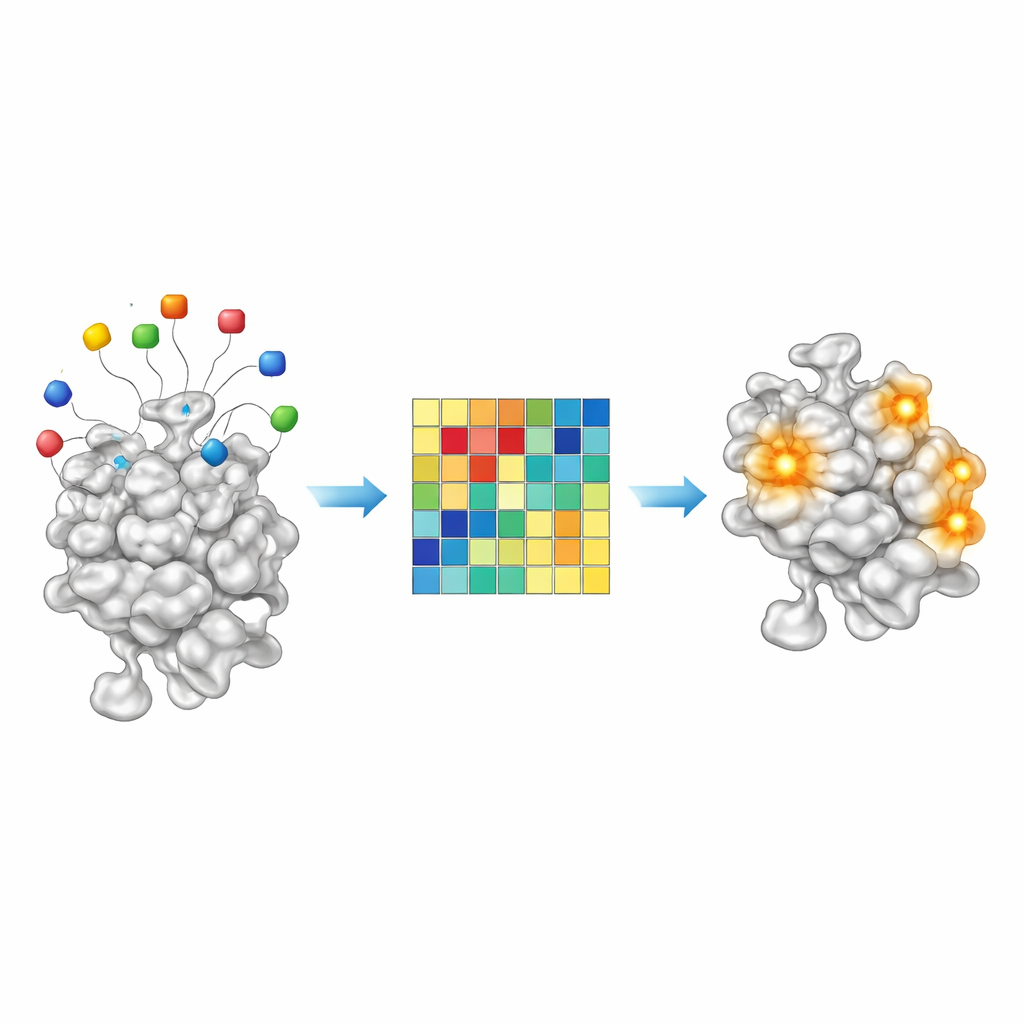
Scanner le protéome humain à la recherche de nouvelles poches
Munis de leur modèle entraîné, l’équipe a ensuite appliqué AF2BIND aux structures prédites par AlphaFold de l’ensemble du protéome humain. Après avoir élagué les régions de faible confiance et découpé les protéines très grandes en fragments structuraux gérables, ils ont regroupé des résidus voisins à score élevé en sites de liaison candidats. AF2BIND a prédit plus de 20 000 de ces sites dans plus de 13 000 protéines. Fait remarquable, la majorité ne chevauchait pas les poches déduites par des méthodes basées sur l’homologie comme AlphaFill, qui copient des ligands depuis des structures cristallines apparentées, ni celles détectées par un chercheur de poches largement utilisé, P2Rank. Beaucoup de sites identifiés uniquement par AF2BIND sont moins profonds ou plus diffus que les poches enterrées classiques et coïncident souvent avec des régions qui lient des peptides, ARN, ADN ou d’autres protéines — des interfaces qui peuvent néanmoins être ciblables par de petites molécules.
Implications pour la découverte de médicaments et les maladies
Pour évaluer le potentiel de ces nouveaux sites pour la conception de médicaments, les auteurs ont utilisé un outil indépendant qui note la « druggabilité » en fonction de la taille de la poche, de son enclosure et de son environnement chimique. En moyenne, les sites d’AF2BIND dépassaient un seuil courant considéré comme attractif pour des cibles médicamenteuses, y compris des sites trouvés dans des protéines liées à des maladies héréditaires. Lors d’un croisement avec des expériences chimio-protéomiques qui marquent des cystéines réactives dans les cellules, AF2BIND et P2Rank expliquaient ensemble près de la moitié des régions détectées comme ligan-dables, chaque méthode repérant des cas que l’autre manquait. Ce travail montre que les représentations internes apprises par les réseaux de prédiction de structure peuvent être réutilisées pour cartographier à grande échelle des sites probables de liaison aux médicaments, sans connaissance préalable d’un ligand spécifique. Pour les non-spécialistes, le message clé est que les mêmes avancées en IA qui prédisent les formes des protéines commencent aussi à révéler où et comment les médicaments pourraient le mieux s’y accrocher, accélérant potentiellement la recherche de nouveaux traitements et éclairant des points de contrôle jusqu’ici cachés dans nos protéines.
Citation: Gazizov, A., Lian, A., Goverde, C. et al. AF2BIND: predicting small-molecule binding sites using the pair representation of AlphaFold2. Nat Methods 23, 626–635 (2026). https://doi.org/10.1038/s41592-026-03011-2
Mots-clés: sites de liaison des protéines, découverte de médicaments, AlphaFold2, biologie computationnelle, bioinformatique structurale