Clear Sky Science · fr
DECODE : cadre de déconvolution commun basé sur l’apprentissage profond pour diverses données omiques
Pourquoi cette recherche est importante
La biomédecine moderne regorge de mesures de nos tissus : quels gènes sont actifs, quelles protéines sont présentes et quelles petites molécules alimentent nos cellules. Pourtant, la plupart de ces mesures sont effectuées sur des échantillons mélangés, où plusieurs types cellulaires coexistent. L’étude à l’origine de DECODE présente un puissant cadre d’intelligence artificielle capable de démêler ces signaux, en identifiant les cellules et états cellulaires présents, y compris à travers des types de données très différents. Cette capacité pourrait accélérer la recherche sur le cancer, l’immunité et les maladies métaboliques tout en tirant un meilleur parti des échantillons de biobanques existants.
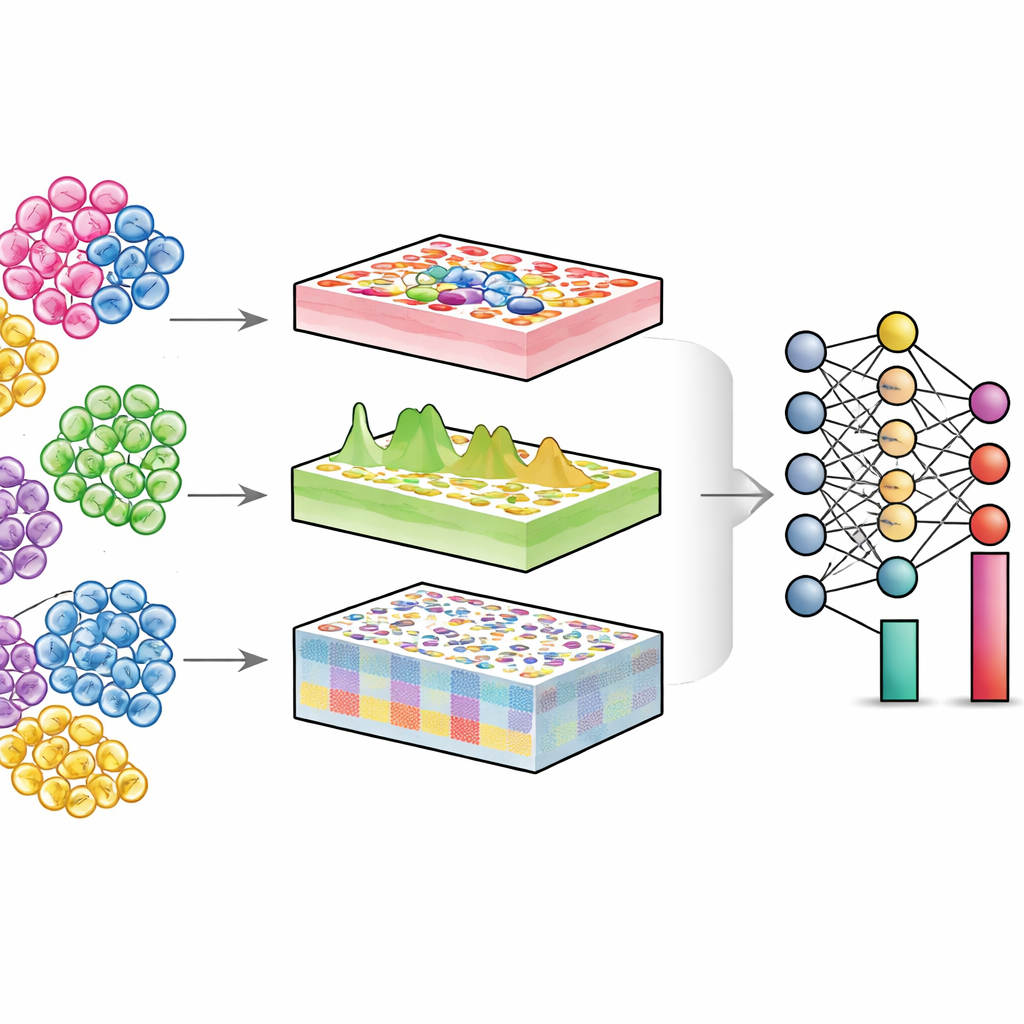
Explorer l’intérieur des tissus mélangés
Chaque organe est une communauté de différents types cellulaires — cellules immunitaires, cellules structurelles, cellules souches, et d’autres. En santé comme en maladie, ce qui change souvent n’est pas seulement la fonction de chaque cellule, mais la proportion de chaque type et leurs états. Les technologies en cellule unique peuvent mesurer les cellules individuellement, mais elles sont coûteuses et techniquement exigeantes, notamment pour de larges cohortes de patients ou des échantillons conservés. En revanche, les expériences « bulk » classiques mixent des milliers ou des millions de cellules et restituer un signal moyen. Les algorithmes de déconvolution cherchent à inverser ce mélange : à partir de données bulk et d’une référence en cellule unique, ils estiment la proportion de chaque type cellulaire dans le tissu.
Les limites des outils mono‑spécifiques
Les outils de déconvolution existants sont généralement adaptés à un seul type de mesure, comme l’activité génique (transcriptomique) ou les protéines (protéomique). Ils supposent souvent des comportements statistiques qui ne s’appliquent pas à d’autres types de données, et peinent lorsque le tissu bulk contient des types cellulaires absents des références. De forts effets de lot — différences liées aux donneurs, instruments ou états de santé — peuvent encore brouiller les signaux biologiques. Notamment, il n’existait pas de méthode pratique pour la métabolomique, l’étude des petites molécules souvent les plus proches des symptômes cliniques. En conséquence, les chercheurs travaillant sur des cohortes multiomiques devaient jongler avec plusieurs outils spécialisés, chacun ayant ses particularités, rendant difficile la comparaison des résultats entre études et types de données.
Un moteur universel de démêlage
DECODE relève ces défis en abordant la déconvolution comme un problème d’apprentissage profond flexible, capable de traiter gènes, protéines et métabolites de façon unifiée. D’abord, il génère des « pseudotissus » en mélangeant numériquement des profils en cellule unique selon des proportions aléatoires, créant un jeu d’entraînement riche où la composition cellulaire vraie est connue. Une étape d’apprentissage adversarial enseigne ensuite à un encodeur à mapper tissus réels et pseudotissus dans une représentation commune où les différences techniques sont minimisées mais les motifs biologiquement pertinents préservés. Ensuite, un module spécial de débruitage, guidé par un apprentissage contrastif, apprend à séparer les véritables signaux tissulaires du bruit artificiel. Cette étape rend DECODE robuste aux types cellulaires manquants dans la référence et aux erreurs de mesure. Enfin, les caractéristiques nettoyées sont transmises à un module de déconvolution qui estime les abondances absolues ou relatives des types et états cellulaires, selon le degré d’exhaustivité de la référence.
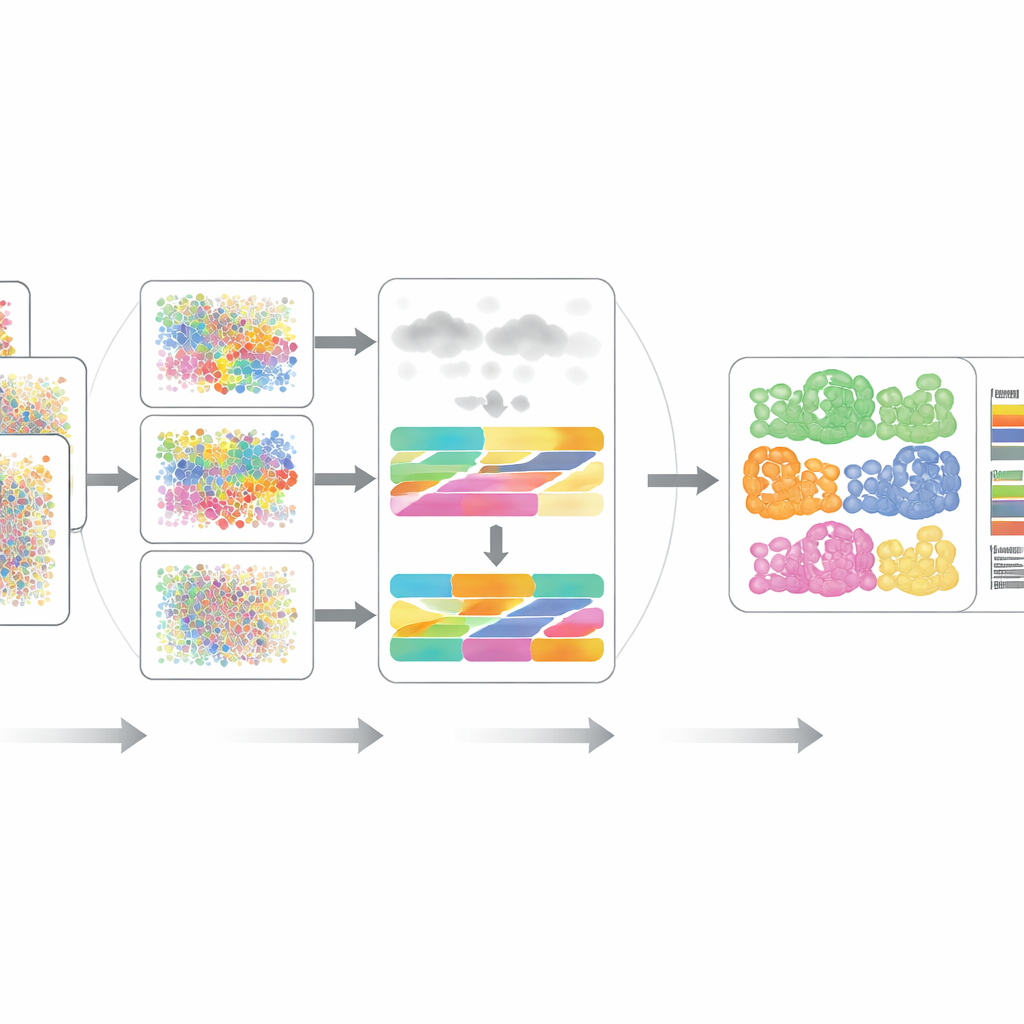
Évaluer DECODE
Les auteurs ont rigoureusement évalué DECODE sur 15 jeux de données couvrant sept scénarios réalistes, incluant différents donneurs, états pathologiques, conditions de santé, plateformes expérimentales et même des mesures à résolution spatiale. Sur la transcriptomique et la protéomique, DECODE égalait ou surpassait généralement les outils de pointe en précision tout en conservant des temps de calcul et une mémoire raisonnables. De manière cruciale, DECODE fut la seule méthode à fournir des résultats fiables en métabolomique, domaine où il y a moins de caractéristiques et où des types cellulaires différents peuvent paraître trompeusement similaires. Le cadre s’est aussi montré apte à suivre des états cellulaires — comme la progression le long d’une trajectoire développementale, les phases du cycle cellulaire ou les réponses à un traitement — plutôt que de se limiter à des types cellulaires statiques.
Robuste face à des données réelles bruyantes et incomplètes
Les tissus réels contiennent souvent des types cellulaires non représentés dans les références en cellule unique en laboratoire, et le bruit expérimental peut altérer simultanément de nombreuses caractéristiques. Les chercheurs ont simulé ces problèmes en ajoutant des types cellulaires inconnus et en introduisant plusieurs formes de bruit et de données manquantes en transcriptomique, protéomique et métabolomique. Dans la plupart des situations, DECODE est resté la méthode la plus précise et, en métabolomique, la seule à ne pas s’effondrer. Ils ont également démontré que DECODE fournit des réponses très cohérentes lorsqu’il est appliqué à des mesures appariées de gènes et de protéines provenant des mêmes échantillons de cellules sanguines, une exigence clé pour comparer les variations de types cellulaires entre couches omiques dans de grandes cohortes.
Nouvelles perspectives biologiques issues de cohortes multiomiques
Grâce à cet outil unifié, l’équipe a réexaminé des jeux de données de maladies complexes. Dans le cancer du sein, ils ont combiné des cohortes transcriptomiques et protéomiques pour montrer comment les cellules immunitaires et les cellules stromales de soutien évoluent entre tumeurs non métastatiques, tumeurs primaires métastatiques et métastases cérébrales. Des motifs tels qu’une abondance plus élevée de lymphocytes T et de cellules de type périvasculaire dans les lésions non métastatiques, et une augmentation des cellules B dans les stades avancés, sont en accord avec et étendent des travaux biologiques antérieurs. Chez la souris, DECODE a intégré des cohortes transcriptomiques, protéomiques et métabolomiques du foie pour suivre comment les hépatocytes, les cellules endothéliales et les cellules immunitaires résidantes changent sous différents régimes alimentaires et modèles de maladie hépatique, reproduisant des tendances connues telles que l’augmentation des fractions de cellules de Kupffer en condition inflammatoire.
Ce que cela implique pour l’avenir
Pour un lecteur non spécialiste, le message principal est que DECODE agit comme un prisme intelligent pour les données biomédicales : à partir de mesures mélangées de tissus, il peut séparer les contributions de nombreux types cellulaires et états, et ce de façon fiable sur plusieurs types de lectures moléculaires. Cela permet aux scientifiques d’extraire beaucoup plus d’informations des cohortes multiomiques et des biobanques existantes sans collecter de nouvelles données en cellule unique pour chaque projet. Bien que la méthode dépende encore de la qualité et de l’étendue des références en cellule unique disponibles, et que les ressources en métabolomique restent limitées, DECODE représente un pas important vers une interprétation systématique au niveau cellulaire des grandes études humaines, avec des bénéfices potentiels pour la compréhension des mécanismes de la maladie et l’orientation de la médecine de précision.
Citation: Zhao, T., Liu, R., Sun, Y. et al. DECODE: deep learning-based common deconvolution framework for various omics data. Nat Methods 23, 596–608 (2026). https://doi.org/10.1038/s41592-026-03007-y
Mots-clés: déconvolution multiomique, référence en cellule unique, apprentissage profond en biologie, analyse métabolomique, composition en types cellulaires