Clear Sky Science · fr
Variation du taux et erreurs récurrentes de séquence en phylogénie à l’échelle pandémique
Pourquoi cela compte pour les prochaines flambées
Lorsqu’un nouveau virus se propage à l’échelle mondiale, les scientifiques se précipitent pour lire son code génétique et reconstruire son arbre généalogique. Ces arbres aident à suivre l’apparition des variants, la vitesse de leur propagation et l’efficacité des mesures de contrôle. Mais pendant la COVID‑19, les laboratoires ont séquencé des millions de génomes de SARS‑CoV‑2 si rapidement que des erreurs cachées et des particularités des données ont commencé à fausser le tableau. Cet article présente de nouvelles méthodes pour nettoyer et interpréter des jeux de données génétiques aussi vastes, offrant une vision plus claire de la façon dont un virus pandémique évolue réellement et circule au sein des populations.
Le défi d’interpréter des millions de génomes
L’épidémiologie génomique transforme les génomes viraux en informations utiles pour la santé publique. Pour le SARS‑CoV‑2, plus de 20 millions de génomes ont été partagés dans le monde. Les outils évolutifs traditionnels ont été conçus pour des problèmes plus modestes, comme la comparaison de gènes entre espèces, et non pour traiter des millions de séquences virales presque identiques arrivant en temps réel. À cette échelle, deux problèmes deviennent particulièrement délicats. D’une part, certains sites du génome viral mutent beaucoup plus souvent que d’autres, ce qui peut faire paraître des virus non apparentés étrangement similaires. D’autre part, des erreurs techniques récurrentes dans le séquençage et le traitement des données peuvent imiter de vraies mutations. Ces deux effets génèrent des « faux échos » dans l’arbre évolutif, créant de l’incertitude sur les branches et regroupements auxquels se fier.
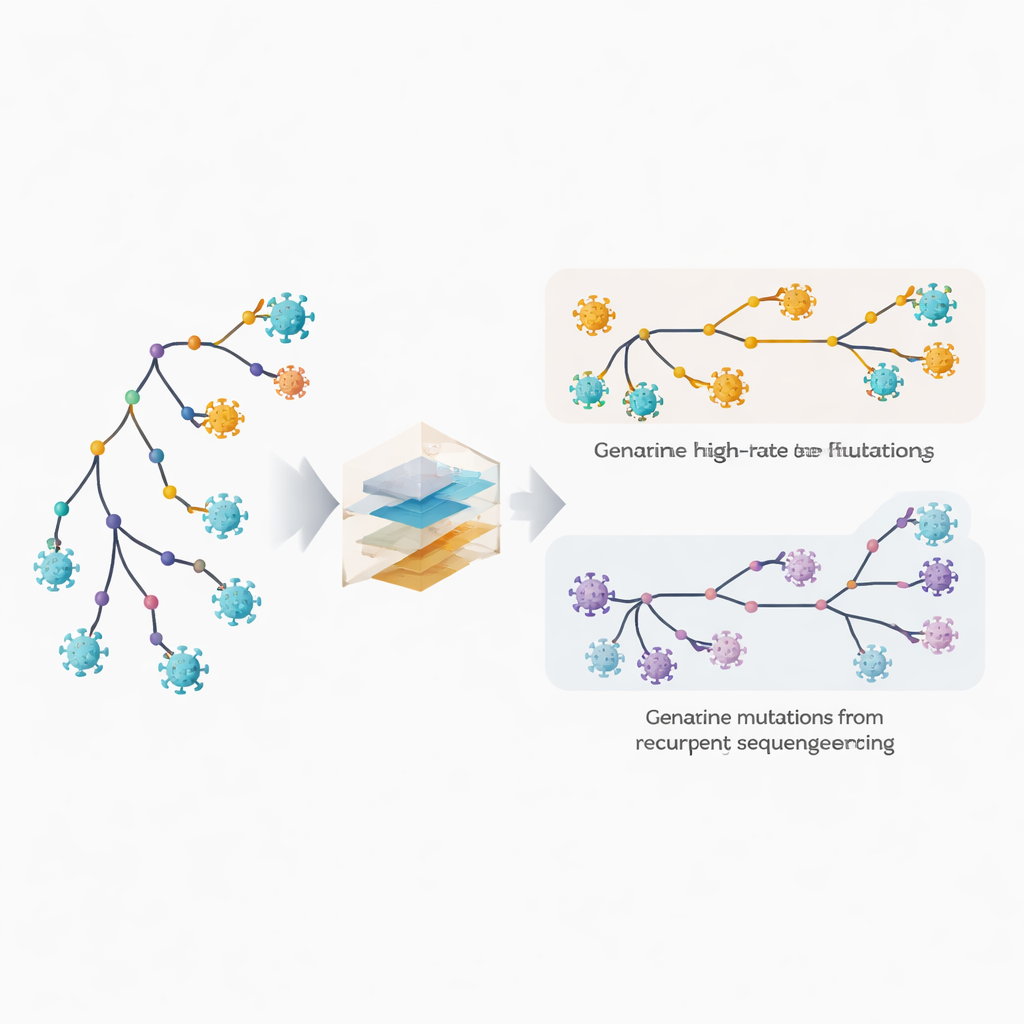
Repérer les sites à évolution rapide et les erreurs cachées
Les auteurs ont étendu leur logiciel phylogénétique, MAPLE, avec des modèles qui traitent chaque position du génome viral comme ayant son propre comportement. Plutôt que de supposer une poignée de taux de mutation moyens, la méthode estime un taux séparé pour chaque site, tirant parti du très grand nombre de génomes disponibles. En parallèle, elle permet à chaque site d’avoir sa probabilité propre de porter une erreur récurrente de séquençage ou d’appel de consensus. L’astuce clé consiste à comparer la fréquence d’apparition d’un changement sur les branches internes profondes de l’arbre, qui reflètent des événements plus anciens et partagés, versus sur les pointes externes, qui correspondent à des génomes individuels. Les vraies mutations biologiques ont tendance à être réparties entre branches internes et terminales, tandis que les erreurs techniques se manifestent principalement aux pointes. En exploitant ce schéma, la méthode peut démêler l’évolution rapide authentique des erreurs répétées.
Algorithmes plus rapides pour un arbre de la vie surchargé
Traiter des millions de génomes nécessiterait normalement une puissance de calcul énorme. Pour rendre l’analyse praticable, l’équipe a repensé la façon dont MAPLE stocke et met à jour l’information séquentielle sur l’arbre. Au lieu de comparer chaque génome à une référence fixe unique, le logiciel sélectionne des « références locales » à l’intérieur de l’arbre et enregistre les génomes proches comme des différences relatives à ces points d’ancrage. Cette représentation compacte accélère les comparaisons entre parties éloignées de l’arbre. D’autres améliorations affinent la manière dont les nouveaux échantillons sont ajoutés à un arbre existant, comment les longueurs de branches sont ajustées, et comment on explore les formes alternatives probables de l’arbre, avec des options pour exécuter les étapes les plus coûteuses en parallèle sur plusieurs cœurs processeur.
Tester la méthode et nettoyer des données réelles
Pour vérifier l’efficacité de leurs modèles, les auteurs ont d’abord créé des jeux de données simulés réalistes de SARS‑CoV‑2 avec des schémas de mutation connus et des erreurs de séquence intégrées. Sur ces tests, la nouvelle approche a reconstruit des arbres évolutifs plus fidèles et localisé des erreurs individuelles avec une grande précision, surtout lorsque des dizaines de milliers de génomes ou plus étaient inclus. Ils se sont ensuite tournés vers des données réelles, analysant des millions de séquences de SARS‑CoV‑2 pour lesquelles les lectures brutes étaient disponibles. En comparant deux pipelines différents de construction de consensus, ils ont repéré des positions génomiques spécifiques affectées de manière répétée par des artefacts, tels que des problèmes de fixation d’amorce ou un biais lié à la référence. Ces sites suspects ont été masqués pour l’analyse ultérieure, et les génomes montrant des signes de contamination ou d’infection mixte ont été filtrés, produisant un alignement épuré de plus de deux millions de séquences de haute qualité.
Un tableau global plus clair de l’arbre généalogique viral
À l’aide du jeu de données nettoyé, les auteurs ont reconstruit un arbre phylogénétique mondial du SARS‑CoV‑2 et cartographié les relations entre les principaux variants. Leur arbre propose parfois des relations légèrement différentes de celles des arbres publics précédents, souvent de façons qui nécessitent moins d’événements de mutation et correspondent mieux au modèle statistique. Le cadre met aussi en évidence les cas où les étiquettes de lignées peuvent être incompatibles avec l’histoire génétique sous‑jacente, signalant des recombinants potentiels ou des génomes problématiques pour un examen plus approfondi. Bien que certains défis subsistent — comme le surapprentissage quand les données sont rares, ou l’influence d’échantillons fortement contaminés — le travail montre qu’il est désormais possible de construire des arbres évolutifs à l’échelle pandémique plus fiables. Pour un lecteur non spécialiste, la conclusion est que mieux gérer les erreurs et les points chauds de mutation permet d’obtenir une vision plus nette de la façon dont les agents pathogènes se propagent et évoluent, aidant les scientifiques et les agences de santé à répondre plus rapidement et avec plus de confiance lors de futures flambées.
Citation: De Maio, N., Willemsen, M., Martin, S. et al. Rate variation and recurrent sequence errors in pandemic-scale phylogenetics. Nat Methods 23, 565–573 (2026). https://doi.org/10.1038/s41592-025-02932-8
Mots-clés: génomique du SARS‑CoV‑2, méthodes phylogénétiques, erreurs de séquençage, variation du taux de mutation, épidémiologie génomique