Clear Sky Science · fr
Modèles d'enantiosélectivité transférables à partir de données éparses
Une manière plus intelligente de trouver le bon catalyseur
Les chimistes cherchent souvent de meilleurs médicaments et matériaux en assemblant des atomes de carbone dans des arrangements tridimensionnels très précis. Obtenir cette subtile différence « main droite » contre « main gauche » — connue sous le nom d'enantiosélectivité — implique généralement d'essayer de nombreux catalyseurs métalliques et conditions de réaction par tâtonnements. Cet article présente une méthode qui utilise des quantités relativement faibles de données expérimentales, combinées à des calculs informatiques rapides, pour prédire quels catalyseurs à base de nickel donneront la chiralité désirée sur une large gamme de réactions, économisant potentiellement des semaines ou des mois de travail de laboratoire.
Pourquoi il est si difficile de contrôler la chiralité des molécules
Beaucoup de médicaments et de produits naturels existent sous des formes images miroir qui peuvent se comporter très différemment dans l'organisme. Les catalyseurs qui favorisent une image miroir plutôt qu'une autre sont donc extrêmement précieux. Mais concevoir de tels catalyseurs est délicat. La chimie quantique traditionnelle peut, en principe, calculer quelle voie une réaction préfère, pourtant de minuscules erreurs d'énergie se traduisent par de grandes erreurs dans la sélectivité prédite, et les calculs sont lents. Les modèles statistiques plus simples, en revanche, sont rapides mais ignorent souvent la danse détaillée entre le catalyseur métallique et les molécules réactives, surtout lorsque le mécanisme réactionnel peut changer subtilement selon les partenaires utilisés.
Capturer les moments clés d'une réaction
Les auteurs comblent cet écart en se concentrant sur les étapes les plus critiques d'une réaction de couplage croisé catalysée par le nickel : les étapes où de nouvelles liaisons carbone–carbone se forment et où le produit final est libéré. Plutôt que d'exécuter des simulations coûteuses de haut niveau, ils utilisent une méthode quantique simplifiée pour générer des structures tridimensionnelles des états de transition et des intermédiaires clés pour de nombreuses combinaisons possibles de catalyseurs et de substrats. À partir de ces structures, ils extraient des centaines de descripteurs physiquement significatifs, tels que l'encombrement autour de certains atomes ou la facilité de déplacement des électrons. Ces nombres sont ensuite injectés dans des modèles de régression linéaire simples qui relient les caractéristiques structurales à la sélectivité mesurée.
Apprendre à partir de données éparses pour guider de nouvelles expériences
Une réalisation centrale de ce travail est qu'il exploite au mieux des données éparses — les combinaisons limitées de catalyseurs et de substrats typiquement rapportées dans un article. Dans une étude de cas, l'équipe revisite une réaction au nickel qui couple des oxydes de styrène avec des iodures aryles. Ils montrent que les descripteurs issus de l'état de transition le plus pertinent surpassent ceux provenant de fragments de catalyseur simplifiés, même si les calculs sous-jacents sont moins coûteux. Avec ces modèles en main, ils testent virtuellement beaucoup plus de ligands sur des paires de substrats existantes et identifient de nouveaux choix de catalyseurs qui augmentent l'excès énantiomérique pour des exemples particulièrement récalcitrants, tout en évitant des dizaines d'expériences inutiles.
Transférer les connaissances entre différentes réactions
Cette approche est puissante parce qu'elle peut être transférée entre des réactions au nickel différentes mais apparentées. Dans un second ensemble d'études, les auteurs combinent des données de plusieurs types de réactions au nickel qui forment toutes des liaisons entre des carbones sp3 et des partenaires comme des groupes aryles ou alkényles, même lorsque les conditions exactes ou les partenaires de couplage diffèrent. En construisant des modèles à partir des mêmes descripteurs mécaniquement significatifs, ils prédisent avec succès l'enantiosélectivité pour de nouveaux ligands, de nouvelles combinaisons de substrats, et même une classe entièrement nouvelle de réaction de formation de liaison carbone–carbone qui n'était pas incluse dans l'ensemble d'entraînement. L'analyse des descripteurs les plus influents suggère aussi quelle étape du cycle catalytique détermine réellement la chiralité pour chaque famille de réactions.
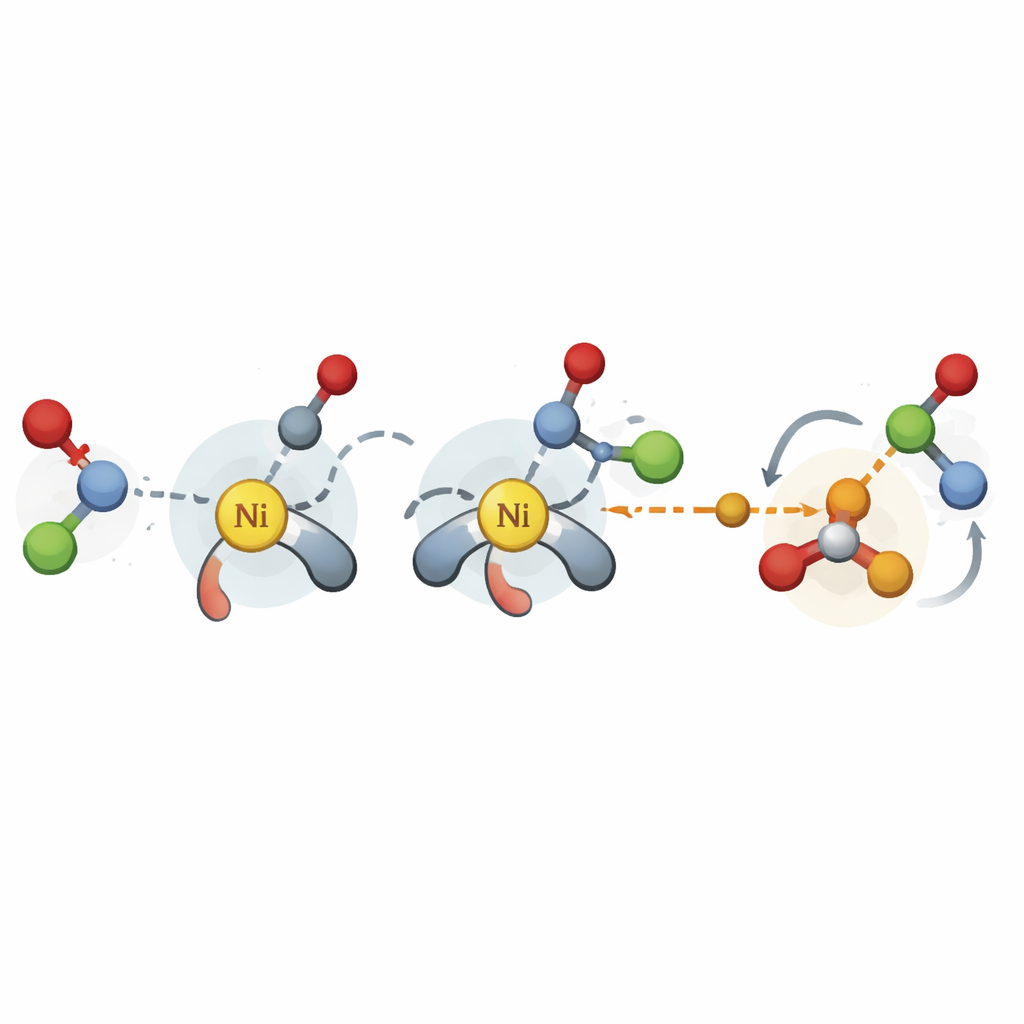
Aider les chimistes à démarrer de nouvelles réactions plus rapidement
Dans une démonstration finale, les auteurs utilisent leur schéma de descripteurs conjointement avec une plateforme d'optimisation bayésienne pour concevoir un couplage catalysé par le nickel entre acétals benzyliques et iodures aryles qui n'avait pas été développé de manière asymétrique auparavant. À partir de données bibliographiques sur d'autres réactions, le modèle recommande de petits ensembles de ligands prometteurs à tester, convergeant rapidement vers la classe la plus performante en seulement quelques dizaines d'expériences. Pour un chimiste, cela signifie un outil pratique pour « démarrer à froid » un nouveau projet catalytique : en alimentant le modèle avec une poignée de résultats initiaux, il peut suggérer quels ligands chiraux sont les plus susceptibles d'offrir une forte enantiosélectivité. Globalement, l'étude montre que des descripteurs choisis avec soin et peu coûteux en calcul peuvent transformer des données passées limitées en indications largement utiles pour concevoir la prochaine génération de réactions sélectives.
Citation: Gallarati, S., Bucci, E.M., Doyle, A.G. et al. Transferable enantioselectivity models from sparse data. Nature 651, 637–646 (2026). https://doi.org/10.1038/s41586-026-10239-7
Mots-clés: catalyse asymétrique, couplage croisé au nickel, apprentissage automatique en chimie, optimisation de réaction, prédiction de l'enantiosélectivité