Clear Sky Science · fr
Les trajectoires d’activation spécifiques aux ligands dictent le signalement des GPCR dans les cellules
Comment un même récepteur peut se comporter comme de nombreux interrupteurs
Beaucoup de médicaments actuels agissent sur une vaste famille de protéines de la surface cellulaire appelées récepteurs couplés aux protéines G, ou GPCR. Ces récepteurs influencent le rythme cardiaque, l’humeur, la respiration et d’innombrables autres fonctions de l’organisme. Pendant des décennies, on les a considérés comme des interrupteurs simples marche/arrêt : un médicament se lie, l’interrupteur bascule et un signal circule à l’intérieur de la cellule. Cet article montre que la réalité est beaucoup plus riche. En utilisant un nouveau type de « mouchard » fluorescent intégré directement dans un récepteur, les auteurs observent, dans des cellules vivantes, comment différents médicaments font parcourir au même récepteur des chemins d’activation distincts — comme choisir des itinéraires différents dans une ville — pour produire des motifs de signalisation différents. Comprendre ces routes cachées pourrait aider à concevoir des médicaments qui apportent le bénéfice souhaité tout en évitant les effets secondaires.
Observer la sonnette d’une cellule en temps réel
On décrit souvent les GPCR comme des sonnettes moléculaires : une molécule de signalisation (un ligand) sonne de l’extérieur, et le récepteur transmet le message aux protéines G à l’intérieur. Des travaux antérieurs sur des récepteurs purifiés en présence de détergents avaient suggéré que les GPCR ne se contentent pas de basculer entre une seule conformation inactive et une seule conformation active. Ils explorent plutôt de nombreuses conformations que les médicaments peuvent stabiliser à des degrés divers. Mais il restait incertain si cette complexité existait aussi dans l’environnement encombré et dynamique d’une cellule vivante. Ici, les auteurs se concentrent sur un récepteur représentatif, le récepteur muscarinique M2 de l’acétylcholine, un régulateur important de l’activité cardiaque et nerveuse, et s’interrogent pour savoir si différents ligands le poussent vers des formes actives distinctes qui ont une importance pour la signalisation cellulaire réelle.
Construire de minuscules témoins lumineux à la surface du récepteur
Pour suivre les mouvements du récepteur sans perturber sa fonction normale, l’équipe a utilisé l’expansion du code génétique, une technologie qui permet d’insérer un acide aminé synthétique spécial à des positions choisies de la surface externe du récepteur. Cette « ancre » chimique peut être cliquée sur un petit colorant fluorescent dans des cellules vivantes. En balayant 72 positions et en ne conservant que celles qui gardaient un comportement comparable à celui des récepteurs normaux, ils ont constitué un panel de sept variantes du récepteur M2, chacune portant un seul colorant sur un site de la boucle externe différent. Lorsqu’on appliquait le messager naturel, l’acétylcholine, la luminosité du colorant à ces sites augmentait ou diminuait de façon caractéristique, révélant comment chaque partie de la surface externe du récepteur se déplaçait au début de la signalisation. De manière cruciale, ces récepteurs marqués pouvaient encore activer les protéines G et subir une internalisation normale, montrant que les témoins étaient fidèles et non perturbateurs.
Les médicaments laissent des « empreintes conformationnelles » distinctes
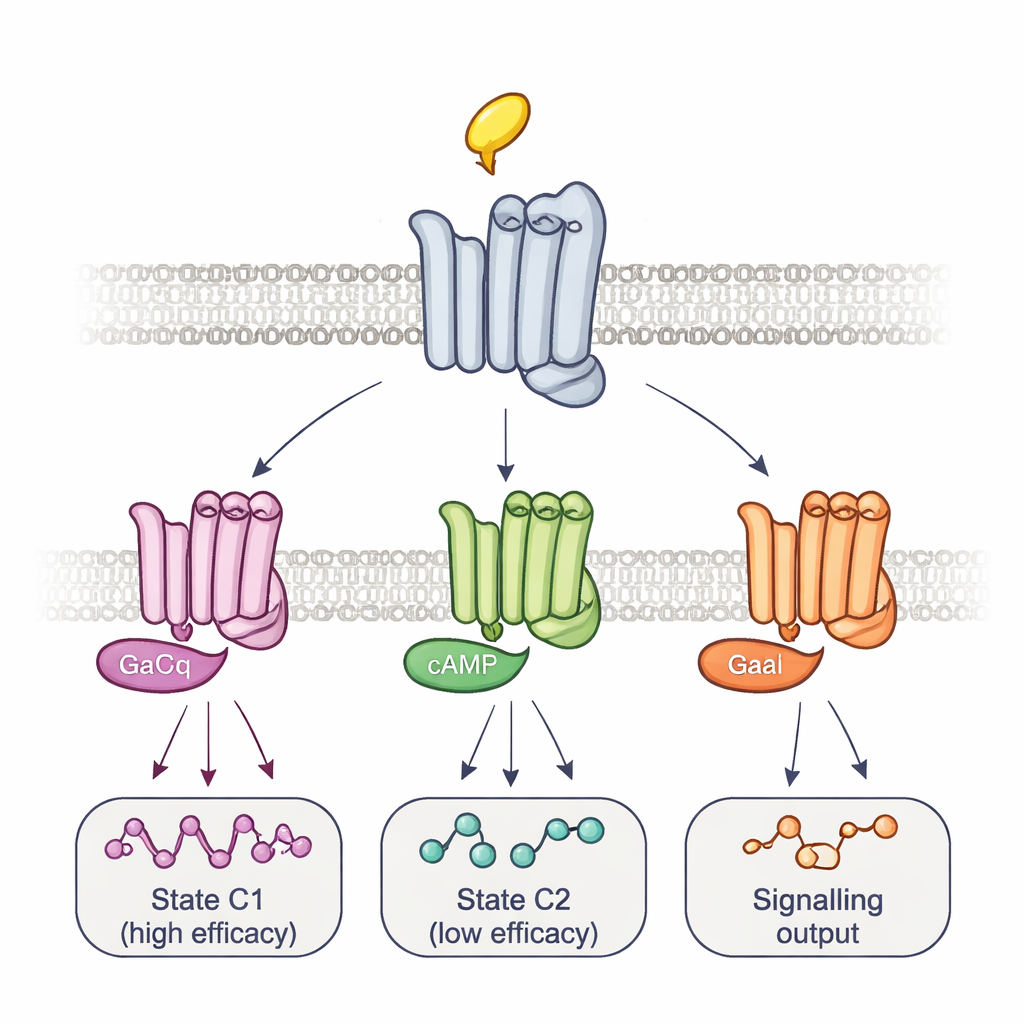
Les chercheurs ont ensuite comparé plusieurs médicaments qui activent tous le récepteur M2 mais avec des intensités différentes : l’acétylcholine endogène, un agoniste synthétique très puissant appelé iperoxo, et deux agonistes partiels plus faibles, l’arécoline et la pilocarpine. Chaque médicament produisait un motif unique de changements de fluorescence à travers les sept sites témoins — une empreinte conformationnelle. À la plupart des positions, l’amplitude du mouvement suivait la force d’activation du médicament. En revanche, à deux sites, la relation était inversée : les médicaments les plus faibles provoquaient les plus grands changements, tandis que le médicament le plus fort n’en provoquait presque aucun. Un tel comportement ne peut pas s’expliquer par un seul état actif. Il indique plutôt que le même récepteur, dans des cellules vivantes, peut adopter plusieurs formes actives distinctes, certaines favorisées par des médicaments puissants, d’autres par des médicaments faibles.
Complexes multiples et itinéraires temporisés vers la signalisation
Pour relier ces conformations à la signalisation réelle, l’équipe a modifié les protéines G elles-mêmes. La surexpression d’une protéine G mutante qui forme des complexes très serrés et de longue durée avec les récepteurs a effacé sélectivement le signal de certains sites témoins tout en en renforçant d’autres. Ce schéma, associé au timing des changements de fluorescence, a révélé au moins deux complexes récepteur–protéine G principaux : un complexe à formation rapide et à haute efficacité et un autre plus lent et moins efficace. Différents médicaments déplaçaient l’équilibre entre ces complexes et utilisaient même des étapes intermédiaires différentes pour y parvenir, retraçant des trajectoires d’activation spécifiques au médicament. À l’aide d’un essai de bioluminescence distinct qui surveille un panel de 14 sous-types de protéines G, les auteurs ont montré que ces équilibres déterminent non seulement l’intensité globale de l’activation par un médicament, mais aussi quels proteins G précis sont activés. Par exemple, l’arécoline activait préférentiellement certains protéines Go, tandis que la pilocarpine favorisait fortement le complexe de faible efficacité.
Pourquoi cela compte pour de meilleurs médicaments
Pour les non-spécialistes, le message clé est qu’un seul récepteur n’est pas un unique interrupteur mais un ensemble d’interrupteurs apparentés, chacun accessible par des chemins différents et chacun connecté à des effets en aval légèrement différents. Cette étude offre une vue directe de ces voies et de ces états dans des cellules intactes, plutôt que dans des systèmes simplifiés en éprouvette. En cartographiant comment des médicaments particuliers biaisent le récepteur vers des complexes et des partenaires protéiques G spécifiques, les chercheurs obtiennent une feuille de route pour concevoir des médicaments « plus intelligents » — des composés qui incitent les récepteurs à adopter des états qui génèrent des signaux utiles tout en évitant ceux liés aux effets indésirables. La stratégie de biosenseur fluorescent développée ici devrait être adaptable à de nombreux autres récepteurs, ouvrant une fenêtre sur la chorégraphie en temps réel de l’action des médicaments dans des cellules vivantes.
Citation: Thomas, R., Jacoby, P.S., De Faveri, C. et al. Ligand-specific activation trajectories dictate GPCR signalling in cells. Nature 650, 1053–1062 (2026). https://doi.org/10.1038/s41586-025-09963-3
Mots-clés: Signalisation des GPCR, Efficacité des ligands, Protéines G, Biosenseurs conformationnels, Découverte de médicaments