Clear Sky Science · fr
Dépistages CRISPR bidirectionnels décodent un circuit cellulaire fibrotique dépendant de GLIS3
Quand la cicatrisation devient nuisible
Nos intestins sont conçus pour se réparer après chaque éraflure ou irritation. Mais dans des maladies chroniques comme la maladie de Crohn et la rectocolite hémorragique, ce processus de réparation peut dérailler, entraînant un tissu cicatriciel épais et rigide qui rétrécit l’intestin et peut nécessiter une chirurgie. Cette étude met au jour une conversation cachée entre cellules immunitaires et cellules structurelles de l’intestin qui pilote cette cicatrisation, et identifie un interrupteur maître, un gène appelé GLIS3, qui pourrait offrir une voie nouvelle pour rompre ce cycle.
Un réseau caché dans des intestins enflammés
Pour comprendre pourquoi certains patients développent une inflammation et une fibrose récalcitrantes, les chercheurs ont construit un « atlas » cellulaire de l’intestin humain. Ils ont combiné le séquençage ARN unicellulaire, qui lit les gènes actifs dans chaque cellule, avec un profilage spatial qui situe ces cellules dans de véritables coupes de tissu. À partir d’échantillons de personnes atteintes de maladie de Crohn, de rectocolite hémorragique et de témoins, ils ont cartographié plus de quatre millions de cellules à travers la paroi intestinale. Parmi cette multitude, un sous-groupe de fibroblastes s’est distingué : les fibroblastes associés à l’inflammation, ou IAF. Ces cellules se rassemblaient dans des zones de colite active et chronique et portaient une signature génique liée à une résistance aux traitements anti‑TNF standards, suggérant qu’elles jouent un rôle central dans les formes difficiles à traiter. 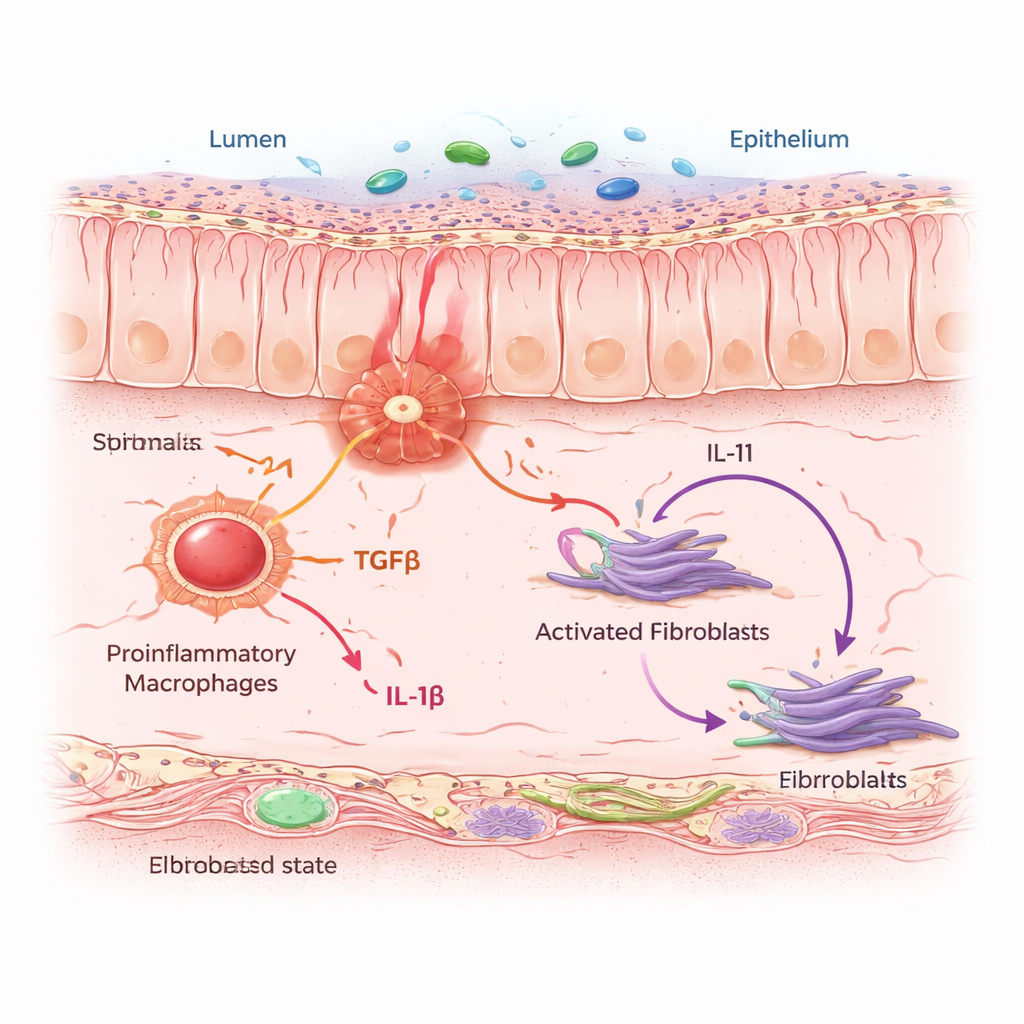
Les macrophages murmurent, les fibroblastes cicatrisent
Les IAF n’agissaient pas seules. Elles se regroupaient en « quartiers » denses en macrophages pro-inflammatoires — des cellules immunitaires qui détectent le danger et libèrent des signaux d’alarme. À l’aide de modèles informatiques et d’expériences de coculture cellulaire, l’équipe a montré que lorsque les macrophages sont poussés dans un état inflammatoire, ils sécrètent deux protéines messagères clés : le TGFβ et l’IL‑1β. Les fibroblastes voisins captent ces signaux via des récepteurs spécifiques. Quand les deux signaux arrivent simultanément, les fibroblastes basculent vers l’état IAF et commencent à produire de l’IL‑11, une cytokine déjà soupçonnée de promouvoir la fibrose, ainsi que du collagène et d’autres protéines de matrice qui épaississent et rigidifient la paroi intestinale. Chez la souris exposée à un protocole de colite chronique, bloquer l’IL‑11 ou la supprimer sélectivement dans les fibroblastes a réduit l’accumulation de collagène sans empêcher l’inflammation initiale, montrant que l’IL‑11 est un moteur crucial de la phase de cicatrisation.
GLIS3 : l’interrupteur maître des fibroblastes fibrosants
Pour passer des corrélations aux mécanismes, les auteurs ont utilisé de puissants outils CRISPR à l’échelle du génome. Ils ont modifié des fibroblastes humains de sorte que la production d’IL‑11 puisse être suivie par une étiquette fluorescente, puis réalisé des criblages parallèles qui désactivaient ou activaient des gènes à travers le génome. En triant les cellules produisant des quantités anormalement élevées ou faibles d’IL‑11 après stimulation au TGFβ et à l’IL‑1β, ils ont identifié des gènes contrôlant cette réponse. Parmi de nombreux composants de signalisation, un facteur de transcription — GLIS3 — est apparu comme un régulateur majeur. Quand GLIS3 était inactivé, les fibroblastes produisaient beaucoup moins d’IL‑11 ; lorsqu’il était renforcé, l’IL‑11 augmentait fortement. Des expériences supplémentaires ont montré que GLIS3 migre dans le noyau des fibroblastes en réponse aux signaux des macrophages, se lie directement à l’ADN près du gène IL11 et d’autres, et active un vaste programme de gènes inflammatoires et fibrosants, y compris des collagènes et des facteurs attirant davantage de cellules immunitaires. 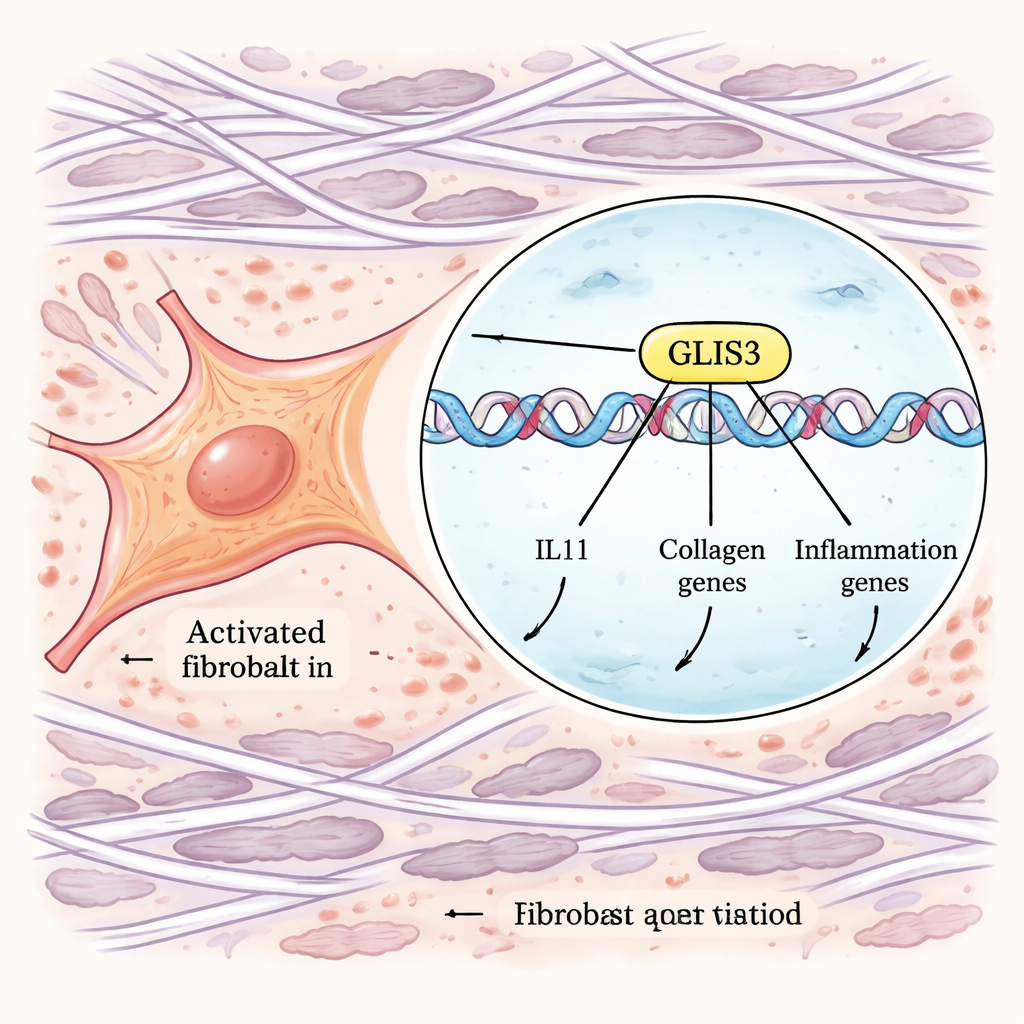
Des modèles murins à la gravité chez les patients
L’équipe s’est ensuite demandé si ce programme piloté par GLIS3 avait une importance in vivo. Chez la souris, ils ont créé une souche où GLIS3 pouvait être supprimé uniquement dans les fibroblastes. Lorsque ces animaux ont été soumis à une colite chronique, ils ont développé moins de cicatrisation intestinale, présenté des niveaux plus faibles de collagène et d’expression de gènes fibrosants, et montré une inflammation réduite comparée aux souris normales. Le cartographage spatial a confirmé que les souris déficientes en GLIS3 comptaient moins de fibroblastes producteurs d’IL‑11 et moins de macrophages activés et de neutrophiles à proximité, indiquant que perturber GLIS3 affaiblit l’ensemble du circuit inflammatoire‑fibrotique. En s’appuyant sur une large cohorte pédiatrique de rectocolite hémorragique, les auteurs ont dérivé une « signature » GLIS3 de 50 gènes et ont constaté que son activité dans des biopsies du côlon suivait de près la sévérité de la maladie et l’abondance d’IAF et de macrophages activés, reliant directement cette voie aux résultats cliniques des patients.
Rompre le cycle de l’inflammation et de la cicatrisation
Pour les non‑spécialistes, la conclusion est que ce travail révèle une boucle autorenforçante : des macrophages inflammatoires poussent les fibroblastes à devenir des IAF producteurs de cicatrices ; ces IAF, sous le contrôle de GLIS3, sécrètent de l’IL‑11, du collagène et d’autres facteurs qui remodelent le tissu et attirent davantage de cellules inflammatoires. Les traitements standards qui suppriment de manière large le système immunitaire peuvent ne pas rompre complètement cette boucle, ce qui aide à expliquer pourquoi de nombreux patients finissent par échouer aux thérapies existantes. En identifiant GLIS3 et l’état fibroblastique producteur d’IL‑11 comme des nœuds centraux du circuit inflammation‑fibrose, cette étude ouvre la voie à des stratégies plus ciblées — visant les fibroblastes plutôt que seulement les cellules immunitaires — qui pourraient un jour prévenir ou inverser la cicatrisation dans les maladies inflammatoires de l’intestin et peut‑être d’autres affections inflammatoires chroniques.
Citation: Pokatayev, V., Jaiswal, A., Shih, A.R. et al. Bidirectional CRISPR screens decode a GLIS3-dependent fibrotic cell circuit. Nature 650, 997–1006 (2026). https://doi.org/10.1038/s41586-025-09907-x
Mots-clés: maladie inflammatoire de l’intestin, fibrose intestinale, fibroblastes, macrophages, GLIS3