Clear Sky Science · fr
L’analyse dynamique des réseaux révèle des couplages de résidus à longue portée à l’interface pMHC sous-tendant une immunogénicité accrue
Comment de minuscules fragments viraux orientent nos défenses immunitaires
Nos cellules T cytotoxiques patrouillent l’organisme à la recherche de signes d’infection ou de cancer. Elles le font en scrutant de petits fragments protéiques, appelés peptides, affichés à la surface des cellules par des molécules connues sous le nom de CMH de classe I. Cette étude pose une question subtile mais importante : comment un simple changement minime dans l’un de ces peptides peut-il rendre la réponse des cellules T beaucoup plus forte — ou complètement absente ? La réponse implique non seulement la structure statique, mais aussi la manière dont l’ensemble moléculaire bouge et se déforme au fil du temps.
La serrure, la clé et les pièces mobiles
Pour comprendre le travail, il est utile d’imaginer le complexe peptide–CMH (pMHC) comme une serrure et le récepteur des cellules T (TCR) comme une clé. Le peptide est logé dans une gouttière du CMH et, ensemble, ils constituent la surface que le TCR explore. Des travaux antérieurs ont montré que la séquence exacte du peptide et la variante de CMH influencent fortement si une cellule T réagit. Les scientifiques ont aussi conçu des « ligands peptidiques altérés » qui portent de petites modifications pour régler les réponses immunitaires, notamment en immunothérapie anti-cancer. Mais alors que nous connaissons bien les formes statiques de ces complexes, nous savons beaucoup moins comment des mouvements en un point du peptide peuvent affecter des régions éloignées de l’interface où le TCR se lie réellement.
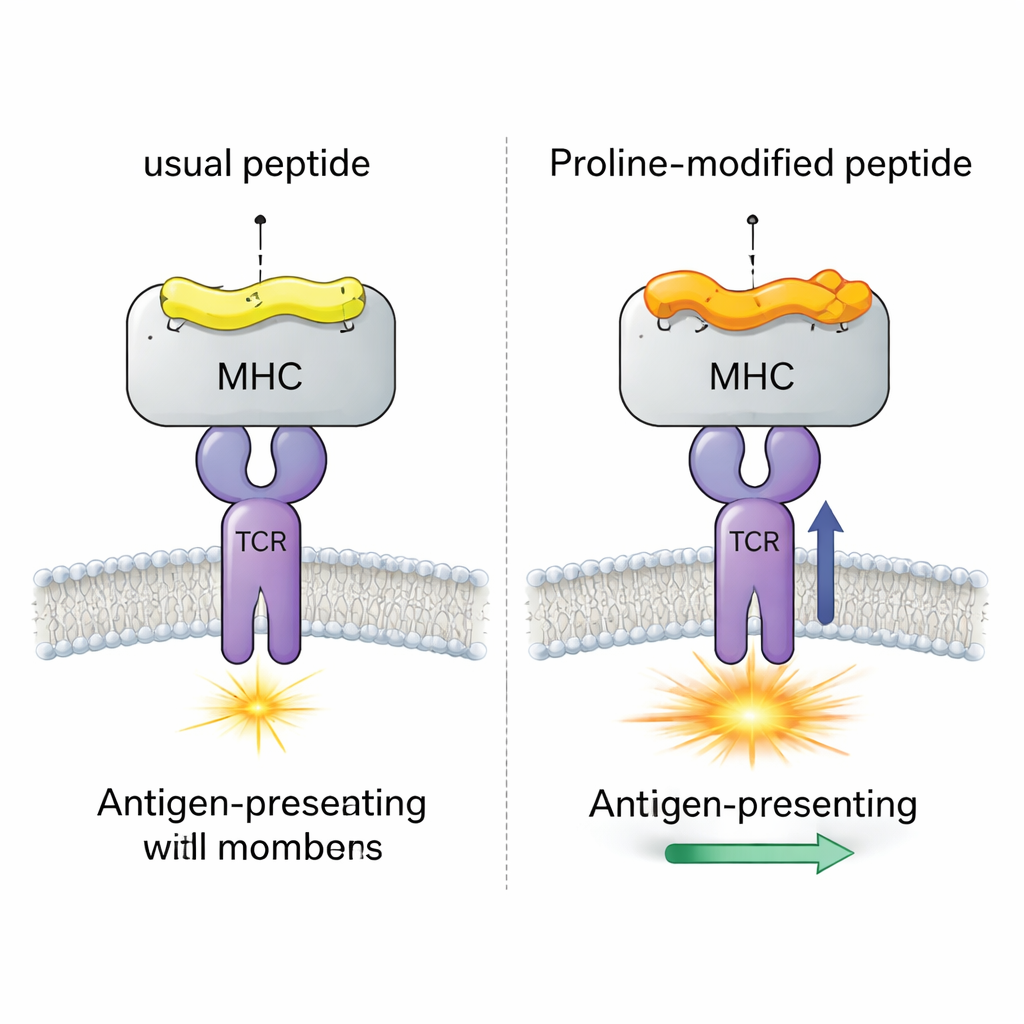
Un cas viral test avec quatre peptides presque identiques
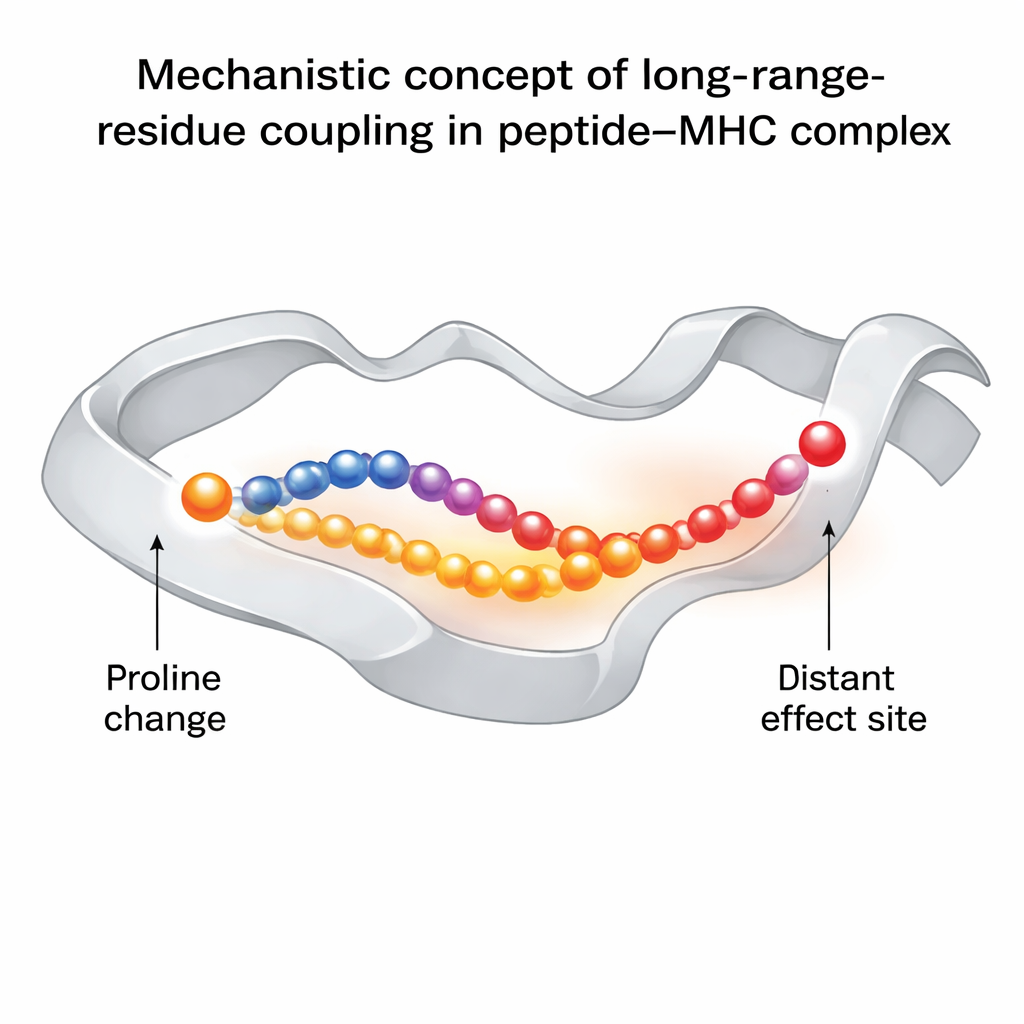
L’équipe s’est concentrée sur un système viral murin bien étudié (LCMV) impliquant le peptide gp33, qui déclenche normalement de fortes réponses des cellules T CD8+. Ils ont comparé quatre versions étroitement apparentées de ce peptide, toutes liées au même CMH (H-2Db). Une version est le peptide viral d’origine ; une autre porte une mutation d’échappement immunitaire que les cellules T reconnaissent à peine ; et deux sont des candidats vaccins « modifiés par proline » où un seul acide aminé près du début du peptide est remplacé par une proline. Des expériences antérieures avaient montré que cet échange pour la proline augmente l’affinité du complexe peptide–CMH et la réactivité d’un TCR modèle (appelé P14), mais le mécanisme détaillé restait flou.
Observer les molécules qui gigotent : simulations et cristallographie
Pour dévoiler ce qui se passe, les auteurs ont combiné des structures cristallographiques haute résolution avec de longues simulations informatiques atome par atome de chaque complexe pMHC en mouvement. Ils ont examiné l’amplitude des fluctuations de chaque résidu au fil du temps et comment ces fluctuations changent lorsque la position trois du peptide est convertie en proline. En corrélant les schémas de mouvement sur de nombreuses simulations appariées, ils ont construit une « carte dynamique » montrant quels résidus bougent de concert, même lorsqu’ils sont éloignés dans l’espace. Ils ont ensuite transformé cette carte en réseau, où chaque résidu est un nœud et les arêtes représentent des mouvements statistiquement liés, et ont analysé ce réseau avec des outils de théorie des graphes similaires à ceux utilisés en analyse de réseaux sociaux.
Communication à longue distance à l’intérieur de la serrure immunitaire
La découverte centrale est que remplacer le troisième résidu du peptide par une proline fait plus que rigidifier localement cette position. Cela modifie la façon dont le mouvement se propage le long d’une des hélices du CMH qui borde la gouttière de liaison au peptide. Cela affecte à son tour le comportement d’un autre résidu du peptide, la position six, qui se situe juste sous l’empreinte du TCR et est critique pour la reconnaissance. Dans les versions « bonnes » modifiées par la proline, ce résidu explore une gamme plus large de conformations, y compris celles optimales pour la liaison au TCR. Dans la variante d’échappement immunitaire sans proline, ce résidu est plus verrouillé et adopte rarement l’orientation favorable au TCR. L’analyse en réseau révèle que cette influence se transmet via des acides aminés spécifiques dans la gouttière du CMH, formant une chaîne de résidus couplés dynamiquement qui relie le site du changement en proline à la région de contact du TCR.
Pourquoi cela compte pour les vaccins et l’immunothérapie
Ces résultats montrent que l’immunogénicité — l’intensité avec laquelle un peptide déclenche les cellules T — n’est pas seulement une question d’ajustement des formes à un instant donné, mais aussi de la manière dont le complexe « respire » et se déforme au fil du temps. Un changement subtil en un point peut se répercuter dans le réseau moléculaire, rendant des résidus de contact clés plus susceptibles de se présenter dans des poses compatibles avec le TCR. Le flux de travail computationnel des auteurs offre une méthode pour détecter systématiquement de tels couplages à longue portée, ce qui pourrait orienter la conception de peptides modifiés pour les vaccins et les thérapies anticancéreuses. En termes simples, ils montrent qu’en choisissant soigneusement où modifier un peptide, on peut pousser la serrure entière vers un état dynamique plus « prêt à être ouvert » par la clé du système immunitaire.
Citation: Resink, T., Sala, B.M., Sun, R. et al. Dynamical network analysis reveals long-range residue couplings at the pMHC interface underlying enhanced immunogenicity. npj Syst Biol Appl 12, 15 (2026). https://doi.org/10.1038/s41540-026-00653-y
Mots-clés: Reconnaissance des cellules T, peptide CMH, dynamique des protéines, ligands peptidiques altérés, immunogénicité