Clear Sky Science · fr
Des variantes du gène MTNAP1 expliquent un trouble neurodégénératif en altérant la stabilité mitochondriale
Pourquoi cette histoire compte pour la santé cérébrale
De nombreuses familles vivent le drame de voir un enfant perdre progressivement des acquis du développement sans diagnostic clair. Cette étude révèle une nouvelle cause génétique de ce type d’affection, reliant un gène défectueux aux « centrales » endommagées à l’intérieur des cellules cérébrales et, en fin de compte, à l’atrophie cérébrale. Comprendre cette chaîne d’événements apporte non seulement des réponses aux familles concernées, mais affine aussi notre compréhension plus large de la vulnérabilité des systèmes énergétiques du cerveau.
Un trouble cérébral infantile nouvellement reconnu
Les chercheurs ont étudié trois enfants issus de deux familles non apparentées, tous présentant des troubles du développement précoces. Ils étaient de petite taille par rapport à leur âge, ont acquis la position assise, la marche et le langage plus tard que prévu, puis ont progressivement perdu certaines de ces aptitudes. Tous ont développé des troubles du mouvement tels qu’une démarche instable, une raideur musculaire ou une hypotonie, et des crises d’épilepsie. Les imageries cérébrales dessinaient un tableau cohérent : les tissus du cerveau (cortex) et du « petit cerveau » à l’arrière (cervelet) s’amincissaient avec le temps, et le corps calleux — un important pont de fibres nerveuses — était anormalement fin. Ces caractéristiques évoquent une perte progressive de neurones plutôt qu’une lésion unique survenue à la naissance.
Un gène minuscule aux grandes conséquences
Pour chercher une cause héréditaire, l’équipe a séquencé tous les gènes codant pour des protéines chez les enfants atteints et leurs parents. Ils se sont focalisés sur un gène nommé MTNAP1, qui contribue à organiser l’ADN à l’intérieur des mitochondries, les centrales énergétiques de la cellule. Chaque enfant portait deux copies défectueuses de MTNAP1, héritées chacune d’un parent sain porteur. Chez deux frères et sœurs, un simple changement d’un « caractère » génétique a remplacé un acide aminé par un autre, déviant subtilement la conformation de la protéine. Chez le troisième enfant, un signal d’arrêt prématuré du gène empêche probablement la synthèse de la protéine. Ces variants n’apparaissaient pas dans de grandes bases de données populationnelles, ce qui renforce l’hypothèse qu’il s’agit de variants rares et délétères plutôt que de simples polymorphismes bénins.
Des centrales énergétiques sous tension
Ensuite, les scientifiques ont examiné des cellules cutanées prélevées chez les enfants et les ont comparées à des cellules de personnes saines. Au microscope, les cellules normales montraient de longues mitochondries filamenteuses formant un réseau connecté, tandis que les cellules des enfants contenaient des mitochondries courtes, fragmentées et agglomérées. Lorsque les chercheurs ont réduit expérimentalement les niveaux de MTNAP1 dans une lignée cellulaire humaine de type nerveux, ils ont observé la même désorganisation du réseau mitochondrial, confirmant que la perte de cette protéine suffit à perturber leur architecture. Les mesures de l’activité mitochondriale ont révélé un affaiblissement d’étapes clés de la production d’énergie, et les cellules produisaient un excès d’espèces réactives de l’oxygène — des sous-produits oxygénés délétères agissant comme une « rouille » moléculaire. Les cellules stressées ont cessé de se diviser correctement, se sont accumulées en phase de repos et ont activé des marqueurs de vieillissement prématuré.
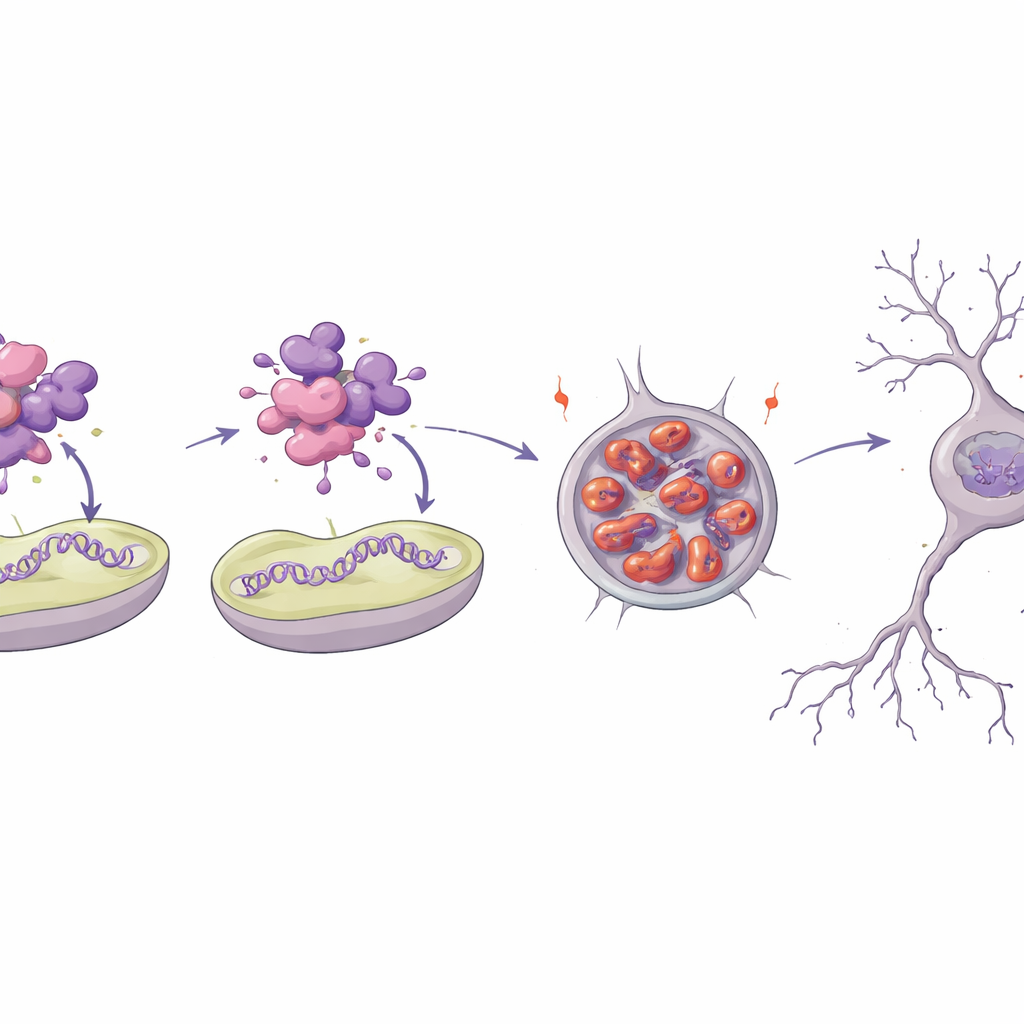
Comment un seul changement défait une protéine vitale
Pour comprendre pourquoi l’un des variants est si perturbant, l’équipe a modélisé la structure 3D de la protéine MTNAP1 et l’a reconstituée en laboratoire. Le résidu substitué se situe dans une région hélicoïdale compactée qui aide normalement la protéine à interagir avec l’ADN mitochondrial et la membrane interne. Des simulations informatiques et des tests biophysiques ont montré que la protéine mutante est moins stable, perd une grande partie de sa structure ordonnée et tend à former des agrégats. Dans des expériences en éprouvette, la protéine normale se liait fortement à de courts segments d’ADN mitochondrial et à des surfaces de membrane artificielles, tandis que la mutant interagissait à peine et s’assemblait plutôt en agrégats de type amyloïde. Lorsqu’elle était introduite dans des cellules de type nerveux, la forme mutante s’accumulait au fil du temps en gros amas péri-nucléaires, signe d’un débordement des systèmes de contrôle de la qualité protéique.
Des mitochondries endommagées à l’échec cérébral
En rassemblant les éléments, l’étude propose un modèle étape par étape : MTNAP1 défectueux affaiblit l’armature qui organise l’ADN mitochondrial et l’ancre à la membrane interne ; cela déstabilise les mitochondries, provoquant leur fragmentation et une perte d’efficacité dans la production d’énergie ; le stress oxydatif croissant et les signaux de vieillissement prématuré rendent alors les neurones particulièrement vulnérables, car ils ont des besoins énergétiques élevés et constants et une capacité limitée à se renouveler. Dans le cerveau en développement, cette crise énergétique lente et persistante se traduit par des retards d’acquisition, une perte de compétences apprises et une atrophie progressive de régions cérébrales clés. Si d’autres patients et des études animales sont nécessaires pour cartographier entièrement le syndrome, ce travail place fermement MTNAP1 comme un gardien crucial de la stabilité mitochondriale et souligne l’organisation de l’ADN mitochondrial comme un pilier central du développement cérébral sain.
Citation: Kumar, A., Saha, S., Nasir, N. et al. Variants in MTNAP1 underlie a neurodegenerative disorder by impairing mitochondrial stability. npj Genom. Med. 11, 19 (2026). https://doi.org/10.1038/s41525-026-00554-3
Mots-clés: mitochondries, neurodégénérescence, génétique pédiatrique, ADN mitochondrial, mauvais repliement des protéines