Clear Sky Science · fr
Potentiels par apprentissage actif pour diagrammes de phases de premiers principes via l’échantillonnage emboîté à échange de réplicas
Pourquoi cela compte pour les matériaux de demain
Des processeurs plus rapides aux pièces d’avions plus résistantes, de nombreuses technologies modernes dépendent de la connaissance des transformations d’un matériau lorsqu’il est chauffé ou soumis à la pression. Ces changements, appelés transitions de phase, sont résumés dans des diagrammes de phases — des cartes indiquant aux scientifiques quelle forme d’un matériau est stable dans quelles conditions. Cette étude présente une nouvelle méthode pour tracer automatiquement ces cartes directement à partir de calculs quantiques, en utilisant l’intelligence artificielle pour réduire drastiquement le coût tout en conservant une grande précision.
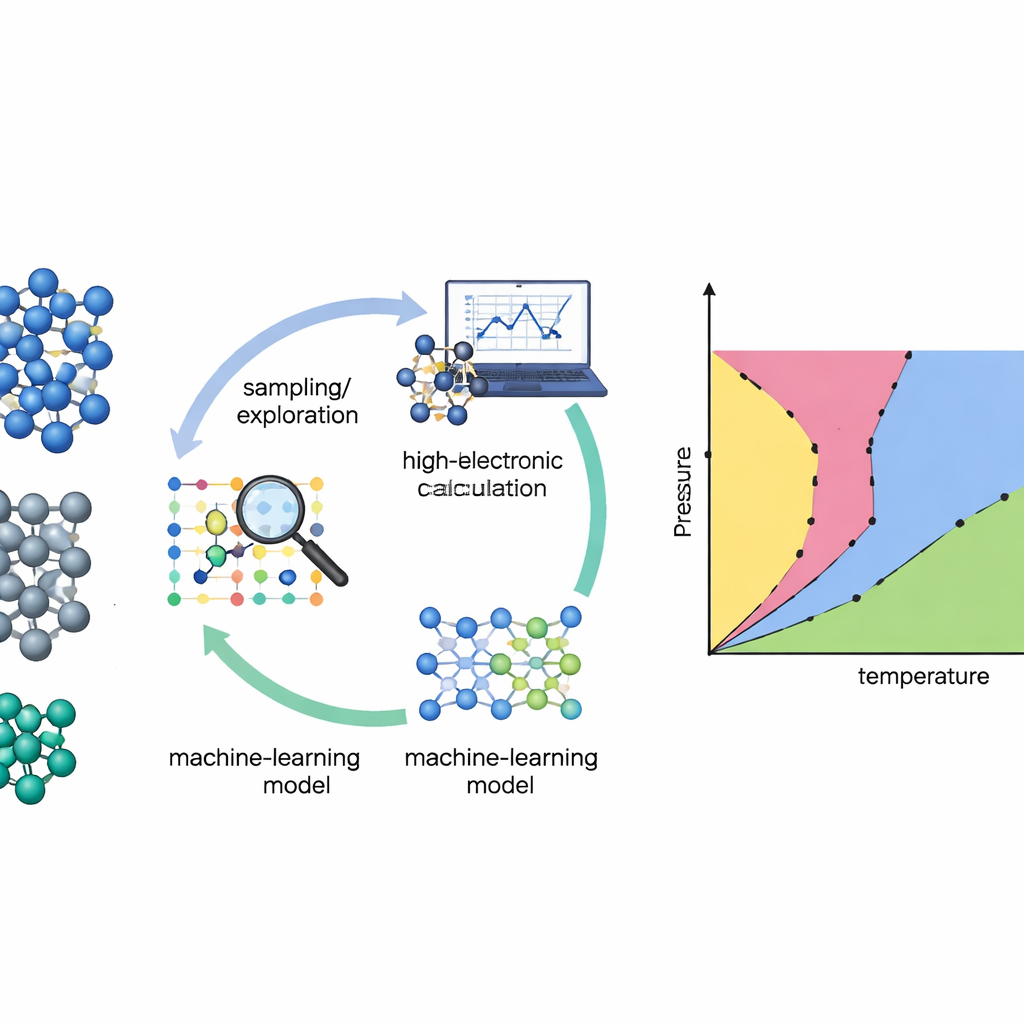
Cartographier les matériaux sans conjectures
Traditionnellement, construire un diagramme de phases à partir des premiers principes revient à randonner dans un paysage accidenté dans l’obscurité : il faut déjà soupçonner où se trouvent les vallées et les cols importants. De nombreuses méthodes standard ne fonctionnent que si les chercheurs fournissent des connaissances a priori solides sur quelles structures cristallines ou « voies » explorer. Les auteurs s’appuient plutôt sur une technique appelée échantillonnage emboîté, qui balaie systématiquement l’ensemble du paysage énergétique d’un matériau sans présumer quelles phases apparaîtront. En suivant l’accessibilité des différentes régions de ce paysage, l’échantillonnage emboîté peut retrouver des propriétés thermodynamiques et des transitions de phase sur une large gamme de températures en une seule passe.
Laisser le modèle choisir ce qu’il doit apprendre
Même la méthode de recherche la plus sophistiquée a besoin d’une bonne description des interactions atomiques. Les calculs quantiques directs (théorie de la fonctionnelle de la densité) sont précis mais trop coûteux pour être évalués des millions ou des milliards de fois. L’équipe résout cela en entraînant des potentiels interatomiques par apprentissage automatique — des modèles rapides qui imitent les forces quantiques entre atomes. Le problème est que ces modèles ne sont fiables que là où ils ont vu suffisamment d’exemples. Pour y remédier, les auteurs construisent une boucle d’apprentissage actif : le modèle d’apprentissage automatique pilote la simulation d’échantillonnage emboîté, signale les configurations où il est incertain, puis demande des calculs quantiques de haut niveau uniquement sur ce sous-ensemble soigneusement choisi. Les nouvelles données sont réinjectées dans le modèle, qui devient plus fiable dans les régions les plus importantes pour le diagramme de phases.
Un nouveau moteur pour explorer le silicium, le germanium et le titane
Les chercheurs ont testé leur approche sur trois éléments importants : le silicium et le germanium, semiconducteurs bien connus, et le titane, un métal structurel largement utilisé. Ils sont partis de bases de données initiales modestes construites à partir de structures cristallines connues et de simples distorsions, omettant volontairement les liquides et de nombreuses configurations à haute énergie. L’échantillonnage emboîté à échanges de réplicas — plusieurs lancements d’échantillonnage emboîté à différentes pressions pouvant échanger des configurations — a ensuite exploré les paysages énergétiques des matériaux. Après chaque cycle d’exploration, l’algorithme sélectionnait automatiquement des centaines de configurations atomiques représentatives, en pondérant celles où ses prédictions de forces divergeaient le plus au sein d’un comité de modèles de réseaux neuronaux. Celles-ci étaient recalculées avec une méthode quantique de haute précision (r2SCAN) et utilisées pour réentraîner les potentiels avant de lancer le cycle suivant.
De débuts bruités à des cartes de phases fiables
En une dizaine à une quinzaine de cycles d’apprentissage, l’incertitude des modèles a progressivement diminué, en particulier sur les forces qui gouvernent le mouvement atomique. Parallèlement, les trajectoires d’échantillonnage emboîté ont commencé à révéler les contours familiers des diagrammes de phases. Pour le silicium, la méthode a reproduit la structure diamant connue à basse pression, sa phase hexagonale à haute pression, ainsi que le comportement de fusion caractéristique en fonction de la température et de la pression, le tout en bon accord avec les expériences et les simulations antérieures. Le germanium a montré un schéma similaire, avec une phase de type diamant à basse pression cédant la place à une phase métallique à haute pression, bien que la pression de transition exacte ait été légèrement déplacée en raison de l’approximation quantique choisie. Le titane a constitué un test plus difficile : ses phases sont métalliques, structurellement proches et séparées par de faibles différences d’énergie. Même là, la stratégie d’apprentissage actif a capturé la séquence des phases solides et la courbe de fusion, et des vérifications supplémentaires à l’aide des fonctions de distribution radiale ont confirmé l’identité des structures prédites.
Ce que cela signifie pour la conception de nouveaux matériaux
En termes simples, l’étude montre qu’un ordinateur peut désormais s’auto‑apprendre le comportement d’un matériau sur une large plage de températures et de pressions, en interrogeant un « oracle » quantique uniquement lorsque nécessaire. Le moteur d’échantillonnage emboîté à échanges de réplicas garantit une exploration large et sans biais, tandis que la boucle d’apprentissage actif veille à ce que les potentiels d’apprentissage automatique soient précis là où cela compte thermodynamiquement. Bien que le travail actuel se concentre sur trois éléments et une méthode quantique particulière, le cadre est général : il peut être associé à des théories électroniques plus avancées ou à des réseaux neuronaux puissants, et étendu à des alliages ou des composés complexes. À mesure que la puissance de calcul et les algorithmes s’améliorent, ce type de flux de travail autonome pourrait devenir un outil standard pour prédire des diagrammes de phases et guider la découverte de nouveaux matériaux aux propriétés sur mesure.
Citation: Unglert, N., Ketter, M. & Madsen, G.K.H. Active learning potentials for first-principles phase diagrams using replica-exchange nested sampling. npj Comput Mater 12, 107 (2026). https://doi.org/10.1038/s41524-026-01989-z
Mots-clés: diagrammes de phases des matériaux, apprentissage actif, potentiels par apprentissage automatique, échantillonnage emboîté, silicium germanium titane