Clear Sky Science · fr
Mélange spatial efficace et précis de potentiels interatomiques appris par machine pour la science des matériaux
Pourquoi des simulations atomiques plus rapides sont importantes
Concevoir de meilleurs matériaux pour des technologies comme la fusion nucléaire, la microélectronique et les alliages structurels dépend de plus en plus de simulations informatiques qui suivent le mouvement et les interactions des atomes. Les méthodes les plus précises s’inspirent de la physique quantique, mais elles sont si gourmandes en calcul que seules des tailles de système et des échelles temporelles modestes sont praticables. Cet article présente ML‑MIX, une technique et un logiciel qui permettent aux chercheurs de conserver une précision proche de la quantique exactement là où elle est nécessaire, tout en utilisant des modèles plus simples et moins coûteux ailleurs. Le résultat est un gain de vitesse substantiel — souvent un facteur de 4 à 10 — sans perte de fiabilité sur les prédictions physiques clés.
Mélanger une vue détaillée et une vue simple des atomes
Au cœur du travail se trouve une idée simple : chaque atome d’une simulation n’a pas besoin du même niveau d’attention. Les régions où les liaisons s’étirent, se cassent ou se réarrangent — comme les défauts, les surfaces ou les particules implantées — tirent profit des potentiels interatomiques apprentis par machine modernes, qui reproduisent la précision mécanique quantique. Mais les atomes éloignés de ces « points chauds » vibrent principalement autour de positions régulières et peuvent être traités par des modèles beaucoup plus simples. ML‑MIX fournit un moyen de combiner un modèle précis mais coûteux avec un modèle plus léger « bon marché » à l’intérieur d’une même boîte de simulation. Il définit une zone centrale qui utilise le modèle coûteux, une zone tampon environnante où les forces sont soigneusement mélangées entre les modèles, et une zone de masse extérieure qui n’utilise que la description bon marché. 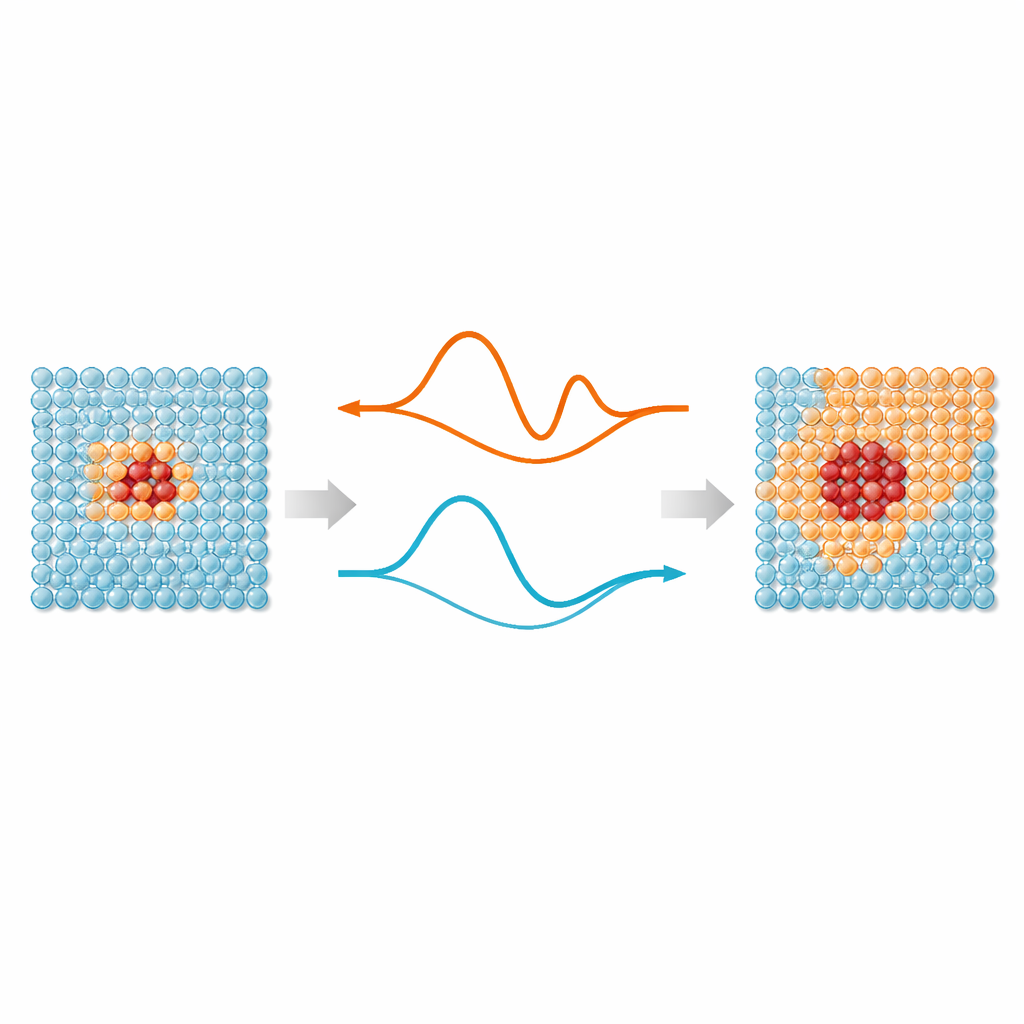
Apprendre au modèle bon marché à imiter le modèle précis
Un défi clé est de s’assurer que le modèle bon marché se comporte comme le modèle précis là où ils se rencontrent. Plutôt que d’ajuster le modèle bon marché directement sur un vaste et varié jeu de données quantique‑mécanique, les auteurs génèrent des données « synthétiques » ciblées en exécutant le modèle précis dans les conditions spécifiques pertinentes pour la région de masse : vibrations à haute température et cristaux légèrement sollicités. Ils ajustent ensuite le modèle bon marché pour qu’il corresponde à ces données, tout en imposant des contraintes strictes sur des propriétés matérielles de base telles que les constantes élastiques et l’espacement du réseau. Cet ajustement contraint garantit que les contraintes et déformations à longue portée correspondent de manière lisse à travers la frontière entre les deux modèles, évitant des forces artificielles qui pourraient corrompre la dynamique près de l’interface.
Mettre la méthode à l’épreuve
Pour vérifier que ML‑MIX fonctionne réellement, les auteurs exécutent une série de tests sur des systèmes de silicium, de fer et de tungstène. Pour un exemple simple, ils calculent la barrière d’énergie pour qu’une vacance — un site vacant du réseau — dans le silicium se déplace d’une position à une autre. La simulation mixte reproduit le résultat d’un calcul entièrement coûteux à moins d’un millième d’électron‑volt, tout en s’exécutant environ cinq fois plus vite. Dans un contexte plus dynamique, ils étirent une seule liaison de silicium dans un cristal chaud et mesurent la force moyenne exercée sur elle. Une simulation n’utilisant que le modèle bon marché s’en tire déjà étonnamment bien, mais dès qu’un petit noyau coûteux est ajouté autour de la liaison étirée, l’accord devient statistiquement indiscernable de la référence entièrement précise, avec des accélérations allant jusqu’à un facteur d’environ 13 en exécutions en série.
Suivre les défauts et les particules en mouvement
Des tests plus réalistes examinent comment les défauts se déplacent dans les métaux. L’équipe simule la diffusion d’un défaut d’auto‑interstitiel dans le fer et d’atomes d’hélium dans le tungstène. Dans chaque cas, le modèle coûteux est confiné à une petite région mobile autour du défaut, tandis que le reste du cristal est traité par le potentiel bon marché. Les coefficients de diffusion résultants correspondent à ceux des simulations entièrement précises dans les erreurs statistiques, même lorsqu’une simulation uniquement bon marché échouerait. Les auteurs poussent ensuite la méthode vers des problèmes plus grands et scientifiquement importants dans le tungstène, un matériau candidat de premier plan pour les réacteurs à fusion. Ils modélisent le mouvement de dislocations en vis — des défauts linéiques qui contrôlent la déformation plastique — et l’implantation d’atomes d’hélium dans une surface de tungstène chaude. Dans les deux cas, ML‑MIX reproduit les résultats issus uniquement du modèle coûteux tout en réduisant le coût de calcul d’un facteur d’environ quatre à onze. 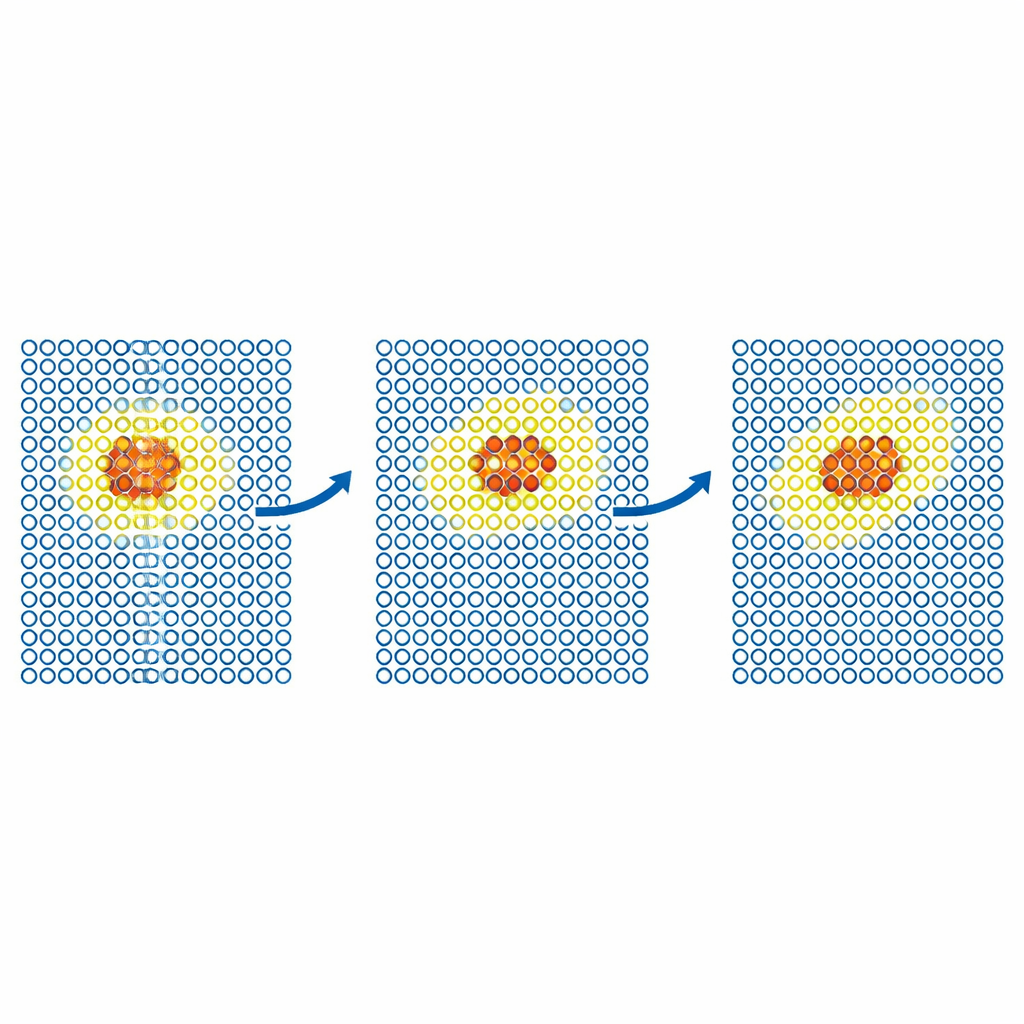
Concilier expériences et perspectives
L’étude sur l’implantation d’hélium illustre le pouvoir de cette approche de manière particulièrement claire. En mélangeant un modèle d’apprentissage automatique de pointe pour les interactions hélium–tungstène avec un potentiel plus rapide pour le tungstène pur, les auteurs simulent beaucoup plus d’événements d’impact et des échantillons de plus grande taille qu’il ne serait autrement possible, le tout sur des processeurs graphiques. La fraction prédite d’atomes d’hélium qui rebondissent sur la surface plutôt que de s’implanter dans le métal concorde avec les mesures expérimentales jusqu’à des énergies d’incidence d’environ 80 électron‑volts, un résultat que les simulations antérieures peinaient à atteindre. Bien que le schéma de mélange ne conserve pas strictement l’énergie et nécessite des thermostats doux, la dérive résultante est faible et gérable. Dans l’ensemble, ML‑MIX démontre que combiner avec soin des modèles atomiques détaillés et simplifiés peut lever des barrières de longue date entre précision et échelle, ouvrant la voie à des simulations de haute fidélité et de routine de matériaux complexes en environnements réalistes.
Citation: Birks, F., Nutter, M., Swinburne, T.D. et al. Efficient and accurate spatial mixing of machine learned interatomic potentials for materials science. npj Comput Mater 12, 110 (2026). https://doi.org/10.1038/s41524-026-01982-6
Mots-clés: potentiels interatomiques appris par machine, simulation multiéchelle des matériaux, implantation d’hélium dans le tungstène, défauts et dislocations, accélération de la dynamique moléculaire