Clear Sky Science · fr
Expansion de grappes atomiques en graphe pour des potentiels interatomiques fondamentaux d'apprentissage automatique
Apprendre aux ordinateurs à ressentir les atomes
Concevoir de nouveaux matériaux pour batteries, avions ou réacteurs à fusion revient souvent à une question simple : comment les atomes se repoussent-ils et s’attirent-ils ? Calculer ces forces exactement est si coûteux qu’une seule simulation peut prendre des jours sur un superordinateur pour un matériau donné. Cet article présente une nouvelle famille de modèles d’apprentissage automatique, appelés GRACE, qui font office de « calculatrice » universelle des forces atomiques sur la plus grande partie du tableau périodique. Leur objectif est de rendre les simulations précises de matériaux complexes courantes plutôt qu’exceptionnelles.
Un seul modèle pour de nombreux matériaux
La plupart des champs de forces appris actuels sont des outils spécialisés : ils fonctionnent très bien pour quelques éléments ou composés, mais doivent être recréés dès qu’on ajoute de nouveaux éléments. GRACE emprunte une voie différente. Il est conçu dès l’origine comme un modèle fondamental capable de traiter 89 éléments chimiques et une très grande variété d’arrangements atomiques avec un ensemble unique de règles. Pour y parvenir, les auteurs s’appuient sur un cadre mathématique appelé expansion de grappes atomiques et l’étendent à des structures de type graphe, ce qui permet au modèle de décrire à la fois les voisinages locaux d’atomes et des motifs plus étendus de manière unifiée. Plutôt que de coder en dur toutes les interactions possibles, GRACE apprend des « embeddings » compacts qui capturent les similarités entre éléments, de sorte que la connaissance d’un matériau puisse aider à décrire un autre.
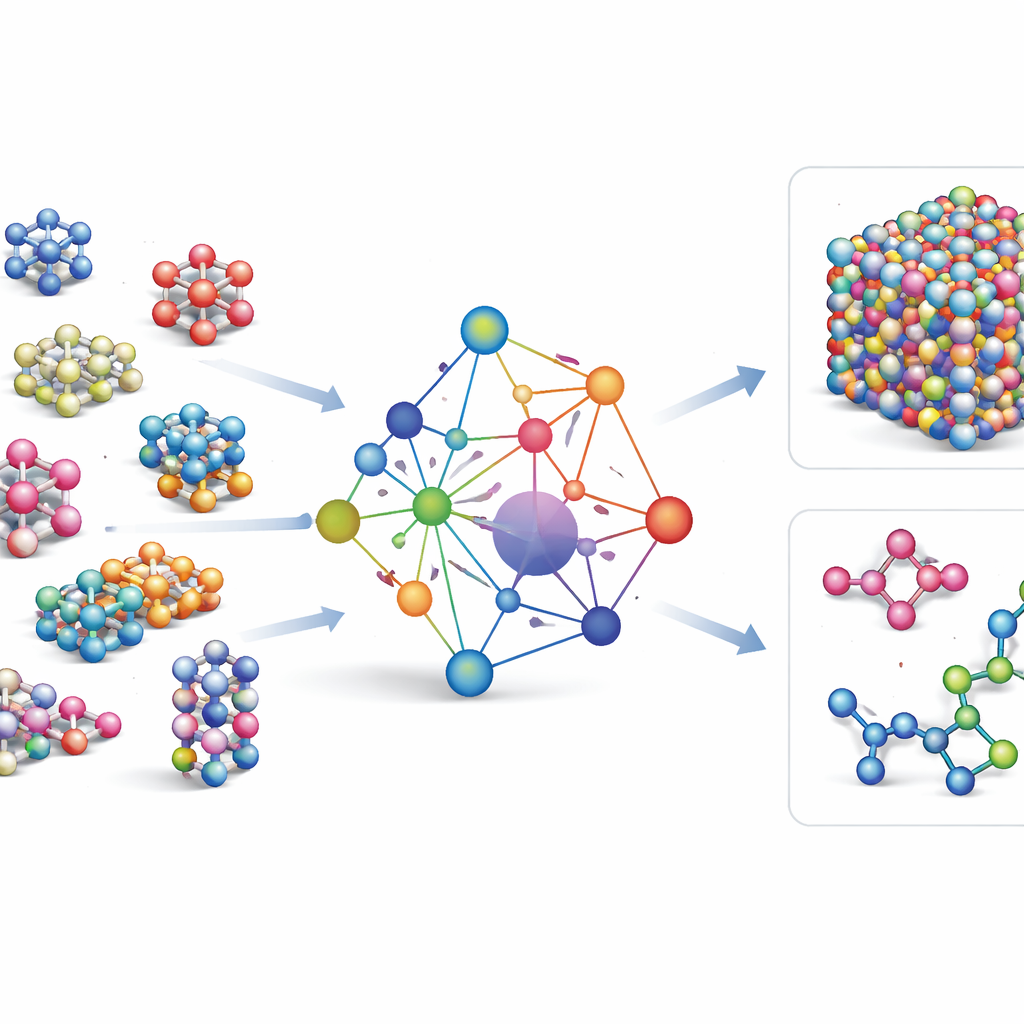
Entraînement sur une mer de données atomiques
Pour apprendre à GRACE le comportement des atomes, les auteurs ont rassemblé certaines des plus grandes bases publiques de calculs quantiques. Le noyau est la collection OMat24, qui contient environ 110 millions de simulations de matériaux inorganiques, complétée par deux autres jeux de données suivant la relaxation et l’évolution des structures. Ensemble, ces ensembles couvrent des cristaux proches de l’équilibre, des structures déformées et contraintes, des liquides à haute température, et plus encore, pour le même large éventail d’éléments. Les modèles GRACE existent en plusieurs tailles, depuis des versions plus simples à une couche qui n’examinent que l’environnement atomique local jusqu’à des versions à deux couches plus profondes qui échangent effectivement des « messages » entre régions voisines. L’entraînement initial vise un bon compromis entre énergies, forces et contraintes internes, et un affinage ultérieur ajuste les modèles pour qu’ils soient compatibles avec des bases de référence largement utilisées en science des matériaux.
Soumettre le modèle à l’épreuve
Un modèle universel n’est utile que s’il fonctionne de manière fiable sur de nombreuses tâches. Les auteurs ont donc soumis GRACE à une batterie de tests exigeante qui reflète l’utilisation réelle des simulations atomistiques par les scientifiques. Sur une référence communautaire pour la découverte de structures cristallines stables, GRACE se situe systématiquement sur le « front de Pareto » : pour une précision donnée, il est plus rapide que les modèles concurrents, et pour une vitesse donnée, il est plus précis. Des avantages similaires apparaissent lors de la prédiction de la conductivité thermique, propriété très sensible aux moindres variations du mouvement atomique. GRACE obtient aussi de bons résultats sur les propriétés élastiques, les énergies de surface, les énergies de joints de grains et les énergies de formation de défauts ponctuels dans de nombreux métaux purs, qui sondent la réponse des matériaux à l’étirement, à la coupe ou aux dommages locaux. Une longue simulation de dynamique moléculaire d’un sel fondu chaud montre que le modèle reste numériquement stable pendant des nanosecondes tout en reproduisant des motifs structuraux détaillés et les taux de diffusion atomique.
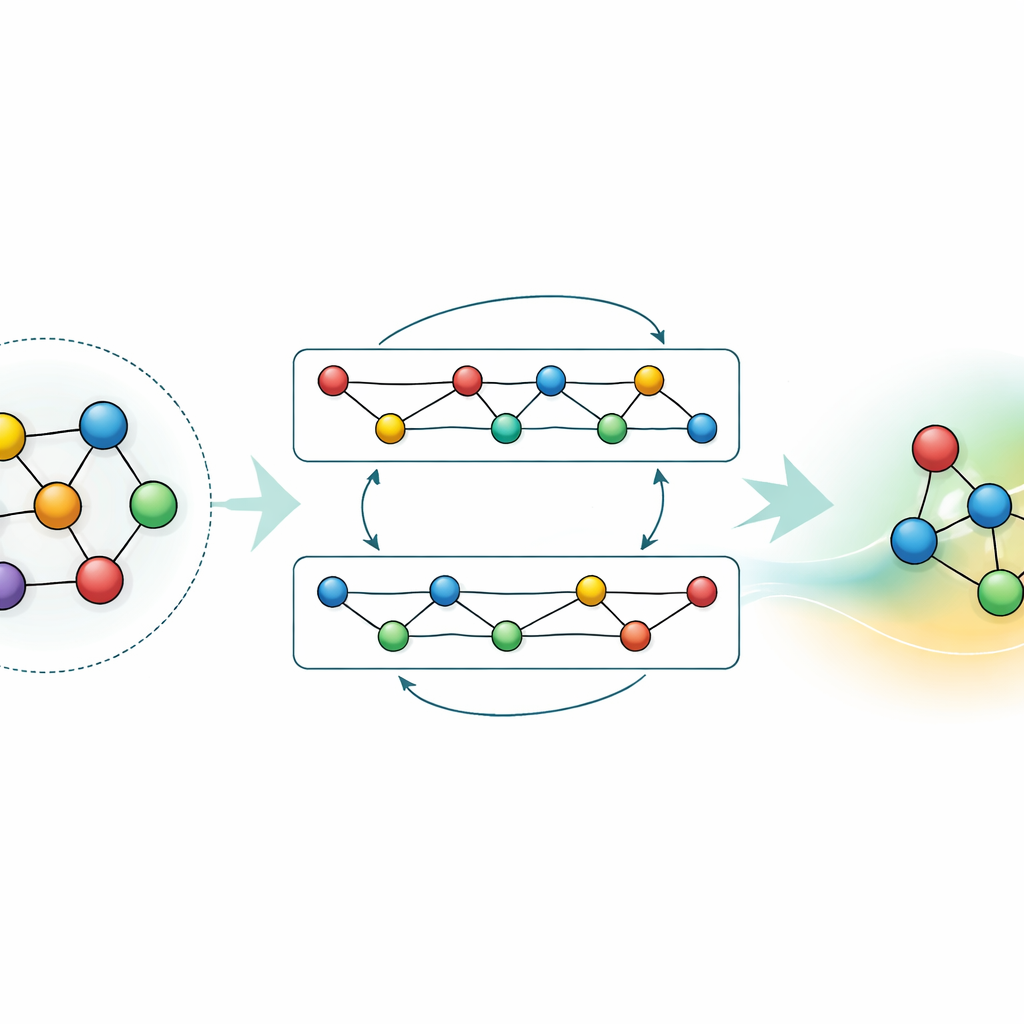
Adapter et compresser les connaissances
Si un modèle polyvalent est puissant, de nombreuses applications exigent soit une précision supérieure pour un matériau spécifique, soit des calculs plus rapides sur du matériel modeste. Les auteurs démontrent deux stratégies pour y parvenir sans perdre ce que GRACE a déjà appris. D’abord, ils affinent le modèle fondamental sur des jeux de données ciblés, comme des alliages aluminium–lithium ou des chemins détaillés de combustion de l’hydrogène. Pour les alliages, même un ajout modeste de données améliore sensiblement les prédictions, surpassant des modèles entraînés de zéro avec la même information. Pour la combustion, un affinage naïf ferait normalement « oublier » au modèle ce qu’il savait d’autres matériaux ; en gelant soigneusement certaines parties du réseau et en ne mettant à jour que des paramètres sélectionnés, les auteurs limitent cet oubli catastrophique tout en gagnant en précision pour la nouvelle chimie. Ensuite, ils montrent comment distiller le grand modèle en un « élève » beaucoup plus simple qui imite le professeur sur des systèmes clés. Cette version distillée s’exécute environ soixante-dix fois plus vite sur CPU tout en conservant la majorité de la précision, surtout lorsqu’elle est entraînée sur un mélange d’alliages complexes et de structures de référence plus simples étiquetées par le GRACE original.
Ce que cela signifie pour la conception future des matériaux
Ce travail positionne GRACE comme une base flexible pour la prochaine génération de modélisation atomistique. Plutôt que de concevoir un nouveau potentiel pour chaque matériau ou propriété, les chercheurs peuvent partir d’un modèle GRACE universel puis l’affiner ou le distiller selon leurs besoins, ce qui économise d’énormes quantités de temps de calcul et de travail d’expert. Les bancs d’essai montrent que cette approche ne se contente pas d’égaler les outils existants ; elle les dépasse souvent en vitesse et en fiabilité, en particulier pour des propriétés exigeantes comme le transport thermique. Pour les non-spécialistes, le message clé est qu’un seul modèle d’apprentissage automatique bien conçu peut désormais servir de « moteur » largement fiable pour des expérimentations virtuelles sur une grande partie du tableau périodique, accélérant la recherche de meilleures batteries, catalyseurs, alliages structurels et matériaux énergétiques.
Citation: Lysogorskiy, Y., Bochkarev, A. & Drautz, R. Graph atomic cluster expansion for foundational machine learning interatomic potentials. npj Comput Mater 12, 114 (2026). https://doi.org/10.1038/s41524-026-01979-1
Mots-clés: potentiels interatomiques par apprentissage automatique, modélisation des matériaux, simulations atomiques, modèles fondamentaux, expansion de grappes atomiques en graphe