Clear Sky Science · fr
Origine des erreurs des champs de forces par apprentissage automatique selon les éléments métalliques
Pourquoi certains métaux sont plus difficiles à « comprendre » pour l’IA
Les modèles d’apprentissage automatique deviennent des outils puissants pour simuler le mouvement des atomes, économisant aux scientifiques d’énormes ressources de calcul par rapport aux calculs quantiques traditionnels. On pourrait s’attendre à ce que les matériaux les plus simples — des métaux purs constitués d’un seul élément — soient les plus faciles à apprendre pour ces modèles. Cette étude montre que ce n’est pas le cas : certains métaux restent obstinément difficiles à décrire, et les auteurs identifient une raison physique à cela.
Construire une grande carte propre du comportement métallique
Pour étudier systématiquement ce problème, les chercheurs ont créé un nouvel ensemble de données appelé Metal-43, basé sur des calculs quantiques exigeants. Il couvre 43 éléments métalliques différents, du lithium léger au tungstène lourd, tous traités avec des réglages de calcul cohérents. Pour chaque métal, ils ont simulé des milliers de structures atomiques à plusieurs températures, enregistrant l’énergie et les forces sur chaque atome. Ce « terrain de jeu » soigneusement contrôlé leur permet de tester des champs de forces par apprentissage automatique — des modèles d’IA qui prédisent les forces atomiques — dans des conditions justes et comparables pour de nombreux métaux. 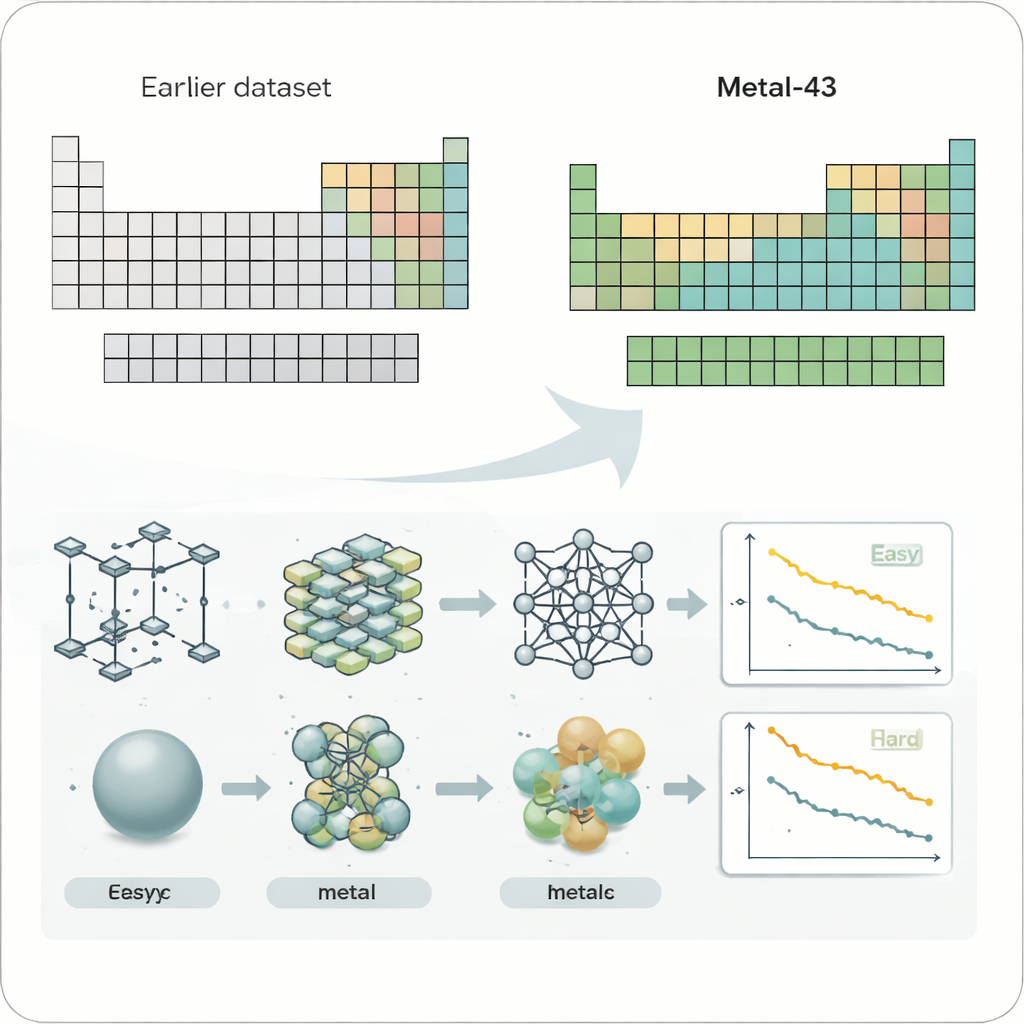
Comment les erreurs des modèles se distribuent dans le tableau périodique
Quatre modèles de champs de forces par apprentissage automatique largement utilisés ont été examinés, incluant à la fois des modèles compacts entraînés séparément pour chaque élément et de grands modèles généraux entraînés sur de nombreux systèmes simultanément. Lorsque les auteurs ont cartographié les erreurs de prédiction sur la disposition du tableau périodique, un motif frappant est apparu. Les métaux mous, faiblement liés comme les alcalins et les alcalino-terreux, ont eu tendance à être plus faciles pour tous les modèles, tandis que les premiers métaux de transition au centre du tableau — souvent utilisés dans les alliages haute performance et les catalyseurs — ont systématiquement produit des erreurs beaucoup plus importantes. Cette tendance est restée valable même lorsque les erreurs brutes ont été re-calibrées pour tenir compte de la force globale des forces atomiques, montrant que la difficulté n’est pas seulement une question de liaisons plus fortes mais qu’il y a quelque chose de plus fondamental.
Complexité cachée dans la « carte de circulation » électronique du métal
L’idée clé du travail est de relier ces erreurs de modèle à la forme de la surface de Fermi de chaque métal, qui est une sorte de « carte de circulation » tridimensionnelle indiquant où les électrons peuvent se déplacer aux énergies les plus pertinentes. Dans les métaux faciles à ajuster, cette surface est lisse et proche d’une sphère. Dans les métaux de transition précoces difficiles, elle devient dentelée et pleine de poches, reflétant un comportement électronique compliqué lié aux orbitales d partiellement remplies. Quand les atomes sont secoués ou légèrement déplacés, ces surfaces de Fermi complexes changent de manière inégale, parfois abrupte, ce qui rend à son tour le paysage énergétique rugueux et complexe. Les auteurs montrent que des mesures numériques simples de la rapidité avec laquelle certaines sommes d’énergies électroniques fluctuent sous de petites perturbations corrèlent fortement avec l’ampleur des erreurs des modèles d’apprentissage automatique, en particulier pour ces métaux de transition problématiques. 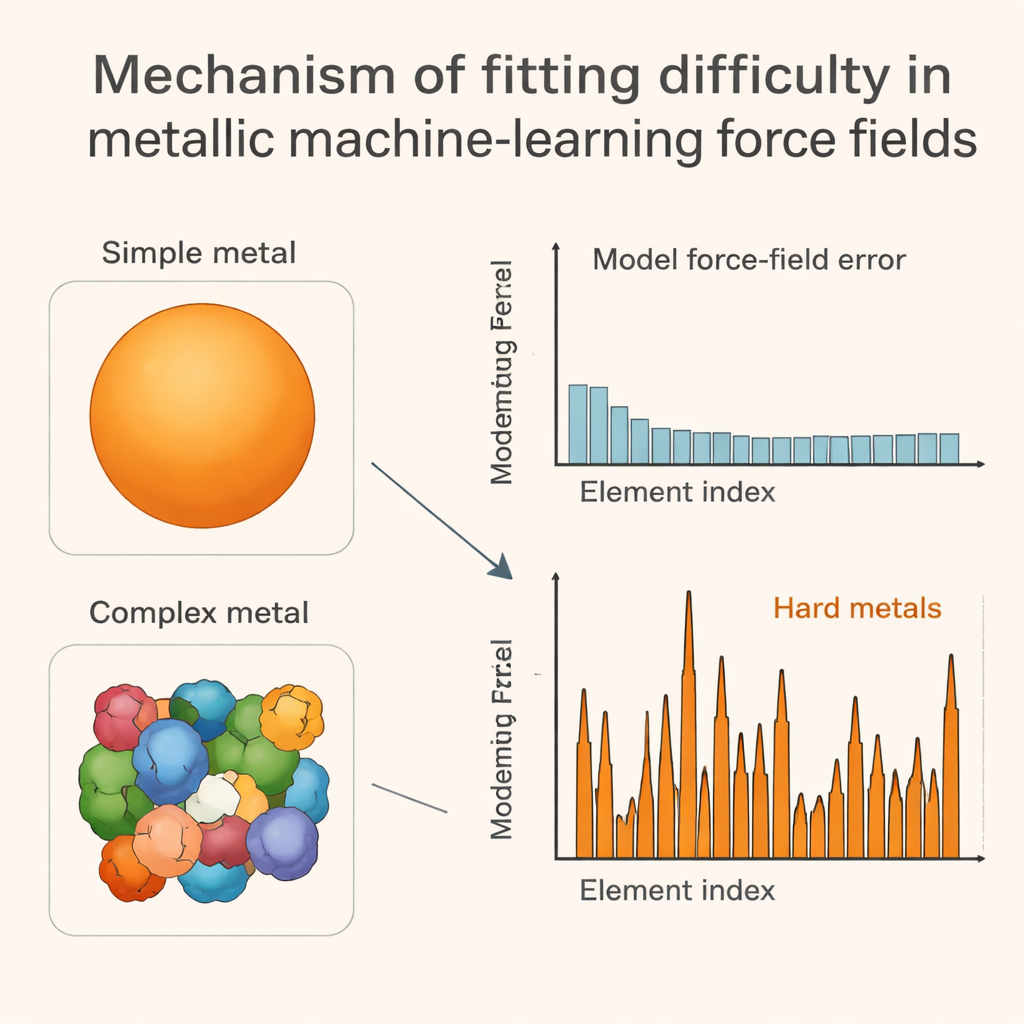
Limites des modèles d’IA actuels, même sur des données idéalisées
Pour dissocier la difficulté inhérente aux métaux des limites des approches d’IA actuelles, l’équipe a aussi généré des jeux de données artificiels en utilisant des modèles traditionnels et faits main des forces atomiques. Certains de ces anciens modèles dépendent principalement des distances entre atomes, tandis que d’autres incluent une forte dépendance angulaire qui imite des liaisons plus directionnelles. Les champs de forces par apprentissage automatique ont pu reproduire presque parfaitement les modèles basés sur la distance, mais leurs erreurs ont augmenté fortement lorsque les effets angulaires étaient importants — en particulier pour les métaux déjà reconnus comme difficiles. Cette comparaison montre que le défi réside non seulement dans la physique sous-jacente des métaux, mais aussi dans le pouvoir de représentation des architectures d’apprentissage automatique actuelles, qui peinent encore avec des interactions many-body fortement dépendantes des angles.
Ce que cela signifie pour les simulations futures
Pour les non-spécialistes, la conclusion principale est qu’il existe une raison claire et physiquement fondée expliquant pourquoi certains métaux sont beaucoup plus durs à modéliser par l’IA que d’autres : la complexité de la mobilité des électrons au niveau de Fermi rend le paysage énergétique rugueux et finement structuré. L’ensemble de données Metal-43 et les indicateurs simples de structure électronique proposés ici offrent aux chercheurs un moyen d’anticiper quels matériaux seront problématiques, d’évaluer équitablement de nouveaux modèles et de concevoir des champs de forces améliorés qui saisissent mieux les liaisons directionnelles. À long terme, ces connaissances devraient aider à rendre les simulations basées sur l’IA plus fiables pour la conception d’alliages avancés, de catalyseurs et d’autres technologies à base de métal.
Citation: Geng, X., Zhang, W., Wang, LW. et al. Origin of the machine learning forces field errors across metal elements. npj Comput Mater 12, 102 (2026). https://doi.org/10.1038/s41524-026-01977-3
Mots-clés: champs de forces par apprentissage automatique, matériaux métalliques, surface de Fermi, potentiels interatomiques, théorie de la fonctionnelle de la densité